Article Text

Abstract
Background Breast cancer (BC) continues to be a major health concern with 250,000 new cases diagnosed annually in the USA, 75% of which are hormone receptor positive (HR+), expressing estrogen receptor alpha (ER) and/or the progesterone receptor (PR). Although ER-targeted therapies are available, 30% of patients will develop resistance, underscoring the need for new non-ER/estrogen-based treatments. Notably, HR+BCs exhibit poor lymphocyte infiltration and contain an immunosuppressive microenvironment, which contributes to the limited efficacy of immunotherapies in HR+BC. In this study, we demonstrate that PR/progesterone signaling reduces major histocompatibility complex (MHC) Class I expression, facilitating immune evasion and escape from immune-based clearance of PR+tumors.
Methods To determine the effect of PR/progesterone on MHC Class I expression, we treated human and mouse mammary tumor cell lines with progesterone and/or interferon (IFN) and measured expression of genes involved in antigen processing and presentation (APP), as well as surface MHC Class I expression. We used the OT-I/SIINFEKL model antigen system to measure the impact of progesterone on immune cell-mediated killing of modified tumor cells. We also analyzed two large BC clinical cohorts to determine how PR expression correlates with APP gene expression and MHC Class I expression in ER-positive tumors.
Results In vitro, we show that PR/progesterone signaling reduces APP gene expression and MHC class I expression in human and breast mammary tumor cell lines. PR-mediated attenuation of APP/MHC Class I expression is more pronounced in the presence of IFN. In immune cell killing assays, PR-expressing mammary tumor cells treated with progesterone are protected from immune-mediated cytotoxicity. We demonstrate that PR expression in vivo prevents immune-mediated rejection of xenoantigen-modified mammary tumor cell lines through mechanisms involving MHC Class I expression and CD8 T cells. Data analysis of two large BC cohorts reveals lower APP gene expression and MHC Class I expression in ER/PR-positive tumors compared with ER-positive/PR-negative tumors. These findings show that HR+BCs, specifically PR+tumors, downregulate APP/MHC class I machinery through PR/progesterone signaling. Use of pharmacological PR/progesterone inhibitors may reverse these effects in patients with BC, thereby improving immunosurveillance and response to immunotherapies.
- Breast Cancer
- Immunosuppression
- Major histocompatibility complex - MHC
Data availability statement
Data sharing not applicable as no data sets generated and/or analyzed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Steroid hormone receptors (HR) such as estrogen receptor (ER) and glucocorticoid receptor (GR) have established roles in regulating antigen processing and presentation (APP) and major histocompatibility complex (MHC) class I expression, which modulate immune surveillance in cancers. Emerging evidence suggests progesterone receptor (PR) may influence tumor immune evasion through modulation of APP/MHC I machinery.
WHAT THIS STUDY ADDS
This study identifies PR as a novel regulator of APP and MHC class I expression in HR+breast cancer (BC). PR signaling via progesterone downregulates APP genes and surface MHC class I expression, facilitating tumor immune evasion by reducing CD8+T cell-mediated killing. Analysis of clinical data sets shows that PR-positive tumors exhibit consistently lower APP and MHC class I expression compared with PR-negative tumors.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
By identifying PR as a critical regulator of APP/MHC I downregulation, this study suggests that targeting PR could enhance immunotherapeutic efficacy in HR+BC. Pharmacological inhibition of PR may restore immune surveillance and sensitize tumors to immune checkpoint blockade, presenting new opportunities for combination therapies aimed at improving outcomes for patients with PR+tumors resistant to current treatments.
Introduction
In the USA 250,000 new breast cancer (BC) cases are diagnosed each year.1 Approximately 75% of these cases are classified as hormone receptor positive (HR+), meaning the tumors express estrogen receptor α (ER) and/or the progesterone receptor (PR).2 3 Despite several classes of drugs that target ER signaling in HR+BC, up to 30% of women develop resistance to these ER/estrogen-targeted therapies and subsequently die from their disease.4 Therefore, novel therapies for HR+BC are urgently needed to improve the health of a significant number of women. Several clinical studies have linked the use of progestins (synthetic progesterone, the native hormone ligand for PR) as part of hormone replacement therapy to an increased incidence of invasive BC,5 suggesting that understanding PR/progesterone signaling in HR+BC is necessary to expand our current treatment options. Previous work from our laboratory demonstrates that PR represses type I interferon (IFN) signaling, a pathway essential for immune-mediated recognition and elimination of developing tumors.6–8 PR-mediated repression occurs through multiple mechanisms: suppression of STAT1 phosphorylation, transcriptional inhibition of interferon-stimulated genes (ISGs), and degradation of STAT2. These processes collectively suppress type I IFN signaling, impairing immune-mediated tumor surveillance. This PR-driven modulation of interferon signaling may contribute to immune evasion, facilitating the development of clinically relevant PR-positive breast tumors.
Major histocompatibility complex (MHC) class I is a crucial molecule expressed on the surface of all nucleated cells. It plays a vital role in relaying information about the health of the cell to the adaptive immune system by binding self/non-self-peptide fragments in the MHC I groove. When a cell becomes virally infected or undergoes malignant transformation, the immunoproteasome cleaves peptides into fragments of 8–12 amino acids in length.9 These fragments are then transported into the endoplasmic reticulum by a heterodimer transporter associated with antigen processing (TAP), TAP 1 and 2 (TAP1 and TAP2). Inside the endoplasmic reticulum, proteases such as endoplasmic reticulum aminopeptidases 1 and 2 further refine the peptides to increase their affinity for the MHC I groove. Once high-affinity peptides bind to MHC class I, the peptide-bound MHC is transported through the Golgi apparatus in secretory vesicles that ultimately end up on the surface of the cell, where it functions to communicate with the immune system (as reviewed in10–12). MHC class I molecules are required for subsequent recognition of cells through the T-cell receptor (TCR) on CD8+T cells, leading to antigen-specific T cell-mediated clearance of the infected/malignant cells. There are several known signaling pathways that regulate the expression of antigen processing and presentation (APP) machinery, including type I and type II IFNs, with type II IFNs (IFN gamma, IFNg) exhibiting the most potent regulatory abilities.13–17 Additionally, DNA damage and NF-κB are also recognized as key regulators of APP machinery.18–21 Given the critical role of MHC class I in effective immune surveillance and immune-mediated tumor control, tumors have evolved numerous mechanisms to downregulate APP expression. These mechanisms include epigenetic silencing of the MHC locus, mutations of the APP machinery genes, or attenuation of the pathways that regulate APP, such as type I and type II IFN (as reviewed in9). The complex regulation of MHC class I expression has important implications for immune function and cancer progression.
Immunotherapies aimed at reactivating exhausted T cells in the tumor microenvironment have revolutionized cancer treatment, but their success has been limited for solid tumors, including mild response rates in endocrine-resistant HR+ metastatic BC.22–26 Although immune checkpoint blockade has shown moderate success in triple-negative BC, it has only yielded mild survival benefits for patients with HR+BC, likely because their tumors have low numbers of tumor-infiltrating lymphocytes (TILs) and weak T cell activation, rendering them less responsive to this immunotherapeutic approach.27 Robust antitumor responses by CD8+T cells require tumor-associated antigens or neoantigens presented on MHC class I. Previous studies have linked steroid HRs, such as glucocorticoid receptor (GR) and ER, to APP/MHC class I downregulation in pancreatic cancer28 and BC, respectively.29 Given the structural similarities between GR and PR, we sought to investigate whether progesterone regulates APP/MHC class I through PR. Moreover, while the studies linking ER to MHC Class I downregulation are provocative, the role for PR as an effector of that downregulation has not been excluded, especially given the link between PR and ER expression. In this study, we show that tumor-intrinsic PR/progesterone signaling downregulates APP/MHC class I machinery and facilitates immune evasion, indicating an important role for PR in tumor development and putatively in the response of HR+BC to immunotherapies.
Material and methods
Cell lines and treatments
T47D non-silencing (NS) and PR short hairpin RNA (shRNA) cells have been previously described.6 MCF7 cells were purchased from American Type Culture Collection, and were grown in Dulbecco’s Modified Eagle Medium (DMEM; Corning) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin. E0771-OVA cells modified to express empty vector or mouse PR (mPR) have been previously described,30 and were cultured in DMEM (Corning) supplemented with 5% FBS and 1% penicillin. Stably transduced pools of E0771-OVA-vec and E0771-OVA-mPR were selected using hygromycin (Thermo Scientific, cat# 10687010) (250 ug/mL) and puromycin (MP Biomedicals, CAS# 58-58-2) (20 mg/mL). E0771-SIINFEKL-vec and mPR-expressing cells were created using the pZIP-MHC-SIINFEKL construct (Addgene) and the expression constructs used to generate the vector and mPR-expressing E0771-OVA cells (constructs referred to below and in30). Lentiviral vectors were produced in an identical manner with OVA vectors described in,30 with cells virally transduced and selected by flow cytometry using BFPTag. Following selection of a high-expressing SIINFEKL population, cells were transduced with the vec or mPR-expressing vectors and stably selected using hygromycin, as below and in30. Flow cytometry confirming SIINFEKL expression, as compared with E0771 cells not expressing the single-chain SIINFEKL construct, is shown in online supplemental figure 1. 67NR cells were modified to express mPR through lentiviral transduction using the pLenti-CMV-Hygro (Addgene #17454, cloning details available on request) backbone. Stably transduced cell lines were selected using hygromycin (250 ug/mL). Resulting stably transduced pools of 67NR-vec and 67NR-PR cells were cultured in DMEM (Corning) supplemented with 10% FBS and 1% penicillin and hygromycin (250 ug/mL). SSM2 were isolated from an ER/PR+mammary gland tumor as previously described31 and were a gift to the Hartman lab from Dr Robert Schreiber (Washington University School of Medicine). SSM2 cells were cultured in DMEM F-12 Nutrient Mixture (Thermo Scientific, #21331020) supplemented with 10% FBS, 2% L-glutamine, 1% penicillin, 0.05 mM b-mercaptoethanol, and HIT supplement (final concentration 0.3 uM hydrocortisone, 10ng transferrin, and 5 ug/mL insulin). Cells were treated with the following reagents where indicated: progesterone/P4 (Sigma-Aldrich, cat # P0130-25G), recombinant mouse IFNg (Invitrogen, cat # BMS326), recombinant human IFNg (BD Biosciences, cat #554617).
Supplemental material
Quantitative RT-PCR
RNA isolation, complementary DNA generation, and quantitative reverse transcription PCR were performed as previously described.7 8 Primers used for quantitative PCR are listed in online supplemental table 1.
Supplemental material
Flow cytometry
Mouse and human cells were seeded at a density of 250,000 cells per well in a 6-well plate, reaching 80% confluency. The cells were then washed with phosphate-buffered saline (PBS) and detached using TrypLE Express (Thermo Scientific). After detachment, the cells underwent two washes with flow buffer (PBS containing 5% FBS) and were subsequently incubated with pan-MHC I antibody (BioLegend clone M1/42 or BD Biosciences Clone: G46 2.6 for mouse or human, respectively) or SIINFEKL-antibody (APC anti-mouse H-2Kb bound to SIINFEKL antibody, BioLegend clone 25-D1.16) on ice, in the dark, for 30 min. Following incubation, the cells were washed twice more with flow buffer. Finally, the expression of MHC I or SIINFEKL was assessed using an Aurora Flow Cytometer (Cytek Biosciences) and FlowJo (BD Biosciences).
67NR tumor growth experiments
67NR-vec or 67NR-mPR cells (1×106 per tumor) were injected into the mammary glands of BALB/c mice (strain #000651; Jackson Laboratory) or SCID-beige mice (strain # CBSCBG-F; Taconic Biosciences). 23 days later, the mice were sacrificed. When applicable, mice were treated with subcutaneous pellet implants (Innovative Research of America) of mifepristone (30 mg/30 days) and onapristone (30 mg/30 days) for 7 days prior to pellet implantation. Mifepristone was obtained from Sigma (M8046), and onapristone was a gift from Context Therapeutics. All mouse experiments were approved by the University of Kansas Medical Center IACUC.
In vivo depletion
To achieve CD8 T cell depletion in BALB/c mice, we administered neutralizing antibodies through intraperitoneal injections. Each mouse received 250 µg of either anti-CD8 antibodies or an isotype control (BioXcell, Clone: 53–5.8 or BioXcell, Clone: HRPN anti-CD8 or Isotype, respectively). The administration schedule was as follows: 3 days prior to tumor injection, and on days 0, 1, 5, 8, 11, 14, 17, and 21 post-tumor injection. On day 5, spleens were analyzed by flow cytometry to verify the specific depletion of CD8 T cells. Control groups were administered equivalent amounts of IgG control.
Splenocyte killing assay
Splenocytes were isolated from control C57BL6/J (strain #000664; Jackson Laboratory) or OTI mice (strain #003831; Jackson Laboratory) and incubated in a 96-well plate with E0771-OVA cells (2,000) at the indicated dilution in RPMI1640/10% Charcoal Stripped FBS/with Pen-Step and L-Glutamine. E0771-OVA cells were modified (as above) to express luciferase to allow for tracking cell viability. Where indicated, media were supplemented with 20 nM progesterone. After 72 hours, non-adherent cells were removed, remaining live cells (attached) were lysed, and luciferase activity was measured (measuring remaining live cells). In all conditions, n=6 biologic replicates and bars indicate SD.
Immunoblotting
Immunoblotting/Western blotting was performed as previously described.8 32 33 Membranes were probed with primary Abs using a PR antibody that detects mPR (Cell Signaling, cat #8757), and β-tubulin (Cell Signaling, cat#2128s).
METABRIC APP gene expression analysis
METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) 34 35 gene expression values were obtained from cBioPortal.36–38 ER-positive tumors with messenger RNA expression were selected for analysis. Gene expression values were taken from the Illumina HT-12 v3 microarray data for genes involved in APP, including: HLA-A, HLA-B, HLA-C, B2M, NLRC5, TAP1, TAP2, TAPBP, PSMB8, and PSMB9. To calculate a score for each tumor, the geometric mean across the APP genes was calculated for each sample. Statistical evaluation of the scores between PR-negative and PR-positive tumors was carried out using a Wilcoxon rank-sum test.
MHC class I expression in the CBCS cohort
Study population: The Carolina Breast Cancer Study (CBCS) is a population-based study conducted in North Carolina (NC) that began in 1993. Briefly, cases of invasive BC between age 20 and 74 years were identified using rapid case ascertainment in cooperation with the NC Central Cancer Registry, with black and young cases (age 20–49 years) oversampled using randomized recruitment. Study details and sampling schemes have been described previously.39 MHC I expression analysis: for this study, we selected 992 patients with HR+/HER2- BC that are classified as PR positive (>10% tumor cells expressing PR) or PR negative (<1% tumor cells expressing PR). Tumor-specific MHC-I (HLA-A/B/C) expression was measured using immunofluorescent assays staining for CK and MHC-I (HLA-A/B/C) on formalin-fixed paraffin-embedded tissue sections. The MHC-I expression data has been previously reported.40 To evaluate the association between PR and MHC-I expression, we performed a two-sample t-test comparing log10 normalized MHC-I fluorescent intensity and the percentage of MHC-I positive tumor cells between PR positive and negative tumors. The statistical analysis was performed in R V.4.1.2.
Statistical analysis
Statistical analysis methods are noted in each figure legend. For tumor growth experiments, differences in tumor growth over time among the groups were analyzed using linear mixed models for repeated measure data using the SAS procedure GLIMMIX (SAS Institute) using AR(1) covariance structure. For all analysis, p≤0.05 is considered significant.
Results
APP gene expression is downregulated by PR/progesterone in BC cells
Previous work from our lab established that PR and progesterone potently attenuated the cellular response to IFNs at multiple steps in the IFN signaling cascade.6–8 30 Importantly, we showed that progesterone-mediated attenuation of IFN signaling leads to transcriptional downregulation of ISGs. Genes in the IFN-signaling pathway aid immune cells in responding to tumors, which includes genes of the APP pathway, leading to upregulation of MHC class I expression to aid in antigen presentation and clearance of tumor cells by CD8+T cells. To determine whether PR signaling reduced the expression of APP genes, we treated the human BC cell line, T47D (ER/PR-positive), with progesterone (or vehicle) for 6 hours, followed by treatment with IFNγ (or vehicle) for 12 hours. Progesterone treatment alone resulted in a modest decrease in the expression of select genes (HLA-A, NLRC5, and TABPB; figure 1A). Interestingly, treatment with progesterone led to a dramatic attenuation of IFNγ-induced APP gene expression of all APP genes assayed, including HLA-A, HLA-B, HLA-C, B2M, PSMB8, PSMB9, TAP1, and TAP2 (figure 1A). While PR signaling appears to modestly downregulate the expression of the APP machinery on its own, pathway-wide attenuation of IFNγ induction is observed for all genes when PR signaling is active. Analysis of publically available Chromatin immunoprecipitation followed by sequencing (ChIP-seq) data sets41 for PR binding in the presence of progestins (progesterone or synthetic progestin, R5020, compared with vehicle control (ethanol; EtOH)) reveals that some of these genes, namely B2M, TAP1, and PSMB9, contain PR binding sites within transcriptional promoters/enhancers (limited to 10 kb up/down from transcriptional start site) regulating APP gene expression (figure 1B). These data suggest that APP gene expression may be regulated directly through PR binding, a finding similar to what we have previously reported regarding PR regulation of IFN-stimulated genes, ISGs.8 Interestingly, TAP1 and PSMB9 are clustered together in one locus (as is observed with many IFN-regulated genes) with a PR binding site detected in a central location to each of these genes. These data suggest that one PR binding event could attenuate transcription of multiple genes involved in APP. Cumulatively, these data show that PR signaling attenuates the ability of IFNγ to transcriptionally upregulate APP gene expression.

PR/progesterone signaling represses interferon response and attenuates APP/MHC class I machinery in human breast cancer cells. (A) T47D ER/PR positive cells were pretreated with progesterone (P4, or vehicle control) at a concentration of 100 nM for 6 hours, then treated with recombinant human interferon gamma (IFN-γ; or vehicle control) at 1,000 U/mL for 12 hours. Following treatment, the cells were harvested, and the gene expression of APP/MHC class I was measured by quantitative PCR (n=3) and normalized to an internal control (B-actin). Error bars represent the SEM between replicates. Statistical differences among the groups were assessed using 2-way ANOVA followed by Tukey’s post hoc tests for pairwise comparisons; relevant comparisons are highlighted. **p≤0.01, ***p≤0.001, **p≤0.01, *p≤0.05. (B) Progesterone-induced PR binding sites in T47D cells shown for APP genes where PR binding was detected within 10 kb of the start site of the gene. ANOVA, analysis of variance; APP, antigen processing and presentation; ER, estrogen receptor; MHC, major histocompatibility complex; mRNA, messenger RNA; PR, progesterone receptor.
MHC class I expression is decreased by PR/progesterone in BC cells
To determine if APP gene downregulation translated to a difference in MHC class I expression, we measured total surface MHC class I expression (HLA-ABC) using flow cytometry in human BC cell lines. Our lab previously generated and characterized a variant of T47D cells stably expressing PR knockdown (T47D PR shRNA), or a NS control (T47D NS shRNA), to correlate gene expression phenotypes directly with PR expression.6–8 42 Treatment with progesterone alone resulted in a modest decrease in surface MHC class I expression, but only in cells with robust PR expression (T47D NS shRNA); PR knockdown cells (PR shRNA) were unresponsive to progesterone treatment (figure 2). When cells were treated with IFNγ ± progesterone, IFNγ-induced MHC Class I expression was downregulated in the presence of progesterone, again only in cells expressing PR (figure 2). Similar phenotypes were observed in a second human ER/PR-positive BC cell line, MCF7 (online supplemental figure 2). These data demonstrate that PR expression and activation leads to downregulation of MHC class I and attenuation of IFNγ-mediated upregulation of MHC class I on the surface of HR+ BC cells.

PR-dependent downregulation of MHC class I expression in breast cancer cells. T47D NS (non-silencing) and PR shRNA cells were treated with progesterone (P4) at a concentration of 100 nM, recombinant human interferon gamma (IFN-γ) at 10 U/mL, or a combination of P4 and IFN-γ for 48 hours. Following treatment, the cells were harvested, and the expression of HLA-ABC was assessed using flow cytometry (histograms on left, quantified on right). Error bars represent the SD between triplicate replicates. Statistical differences among groups were assessed using 2-way ANOVA followed by Tukey’s post hoc tests for pairwise comparisons and relevant differences are highlighted on the graph; **p≤0.01. ANOVA, analysis of variance; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; PR, progesterone receptor; shRNA, short hairpin RNA.
Progesterone-dependent downregulation of MHC class I expression and protection from immune cell-mediated death in mouse mammary tumor cell lines
In order to study the effects of MHC class I downregulation on protection from T cell-mediated tumor cell death, we transitioned our studies to the mouse mammary tumor cell line, E0771, that has previously been engineered to express the model antigen, OVA, and luciferase (to use for tracking cell viability).43 Of note, this cell line, like the vast majority of mouse mammary tumor cell lines, lacks expression of ER and PR.44–46 Therefore, in our previous work, E0771-OVA cells were also engineered to express mouse PR (mPR) or an empty vector,30 in order to study the effects of direct PR action in these tumor lines. Using these cell lines, we observed that treatment of E0771-OVA-mPR cells with progesterone resulted in a decrease in MHC class I expression, while E0771-OVA-vec cells exhibited little change (figure 3). Notably, the combined treatment of progesterone and IFNγ in E0771-OVA-mPR led to lower MHC class I expression compared with IFNγ treatment alone. These phenotypes are most potent in cells expressing PR. E0771-OVA-vec control cells show low levels of responses to progesterone, likely due to their high levels of GR expression (GR can be activated by progesterone); progesterone-mediated phenotypes in E0771 are potentiated in the presence of mPR (E0771-OVA-mPR cells). Importantly, we observed progesterone-dependent downregulation of MHC class I expression in a second mouse mammary tumor cell, SSM2 (online supplemental figure 3), that expresses endogenous PR and ER.31 Cumulatively, these data demonstrate that progesterone/PR activation leads to the downregulation of MHC class I, independent of ER.

PR/progesterone signaling attenuates MHC class I in mouse mammary tumor cells expressing PR. E0771-OVA-vec and E0771-OVA-mPR cells were treated with progesterone (20 nM; P4) or recombinant mouse interferon gamma (IFN-γ; 100 ng/mL). MHC class I expression was assessed using flow cytometry 48 hours post-treatment (histograms on left, quantified on right). Error bars represent the SD between replicates. Fold change between relevant treatment groups is shown in text boxes. Statistical differences among groups were assessed using 2-way ANOVA followed by Tukey’s post hoc tests for pairwise comparisons. All pairwise comparisons are significant (p≤0.05 or less), with the exception of vehicle treatment between cell lines. ANOVA, analysis of variance; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; PR, progesterone receptor.
To investigate the role the immune system plays in controlling PR+mammary gland tumor growth, we performed an immune cell killing assay. For this assay, E0771-OVA control and PR+cell lines were used. These cells had been previously modified to express ovalbumin,43 which permits the presentation of the SIINFEKL peptide by H-2kb, which can be recognized by the TCR from OT-I transgenic CD8+T cells. Additionally, they were modified to express luciferase, to allow for tracking cell viability.43 E0771-OVA-mPR or vector controls were then co-cultured with splenocytes derived from OT-I C57BL/6 mice at various effector to target ratios to elicit tumor cell lysis by OT-I CD8+T cells. Interestingly, E0771-OVA-mPR treated with progesterone were the only experimental group with observed protection from immune cell killing (figure 4A). Protection from immune cell-mediated death was not observed in cells lacking PR expression, indicating that progesterone-mediated protection from death is intrinsic to PR-expressing tumor cells. Additionally, tumor cells were not killed in the presence of splenocytes derived from wt C57BL/6 mice (online supplemental figure 4), indicating that T cell protection conferred be PR/progesterone was antigen-specific. To verify that modulation of MHC Class I expression by PR/progesterone is responsible for protection from immune cell killing, we engineered E0771-vec and mPR cells (lacking OVA) to express a single-chain H-2Kb-SIINFEKL construct (pZIP-MHCI-SIINFEKL, see Methods and online supplemental figure 1). In these cells, SIINFEKL expression, the cognate antigen for OT-I T cells, is driven constitutively from an engineered plasmid and linked to a single-chain MHC Class I gene (with B2M). MHC expression from this construct is not subject to processing and modification through the APP pathway, and should therefore not be susceptible to the progesterone-mediated effects on APP described above. Indeed, in cells constitutively expressing H-2Kb-SIINFEKL, PR expression and progesterone treatment no longer protected E0771 tumor cell variants from immune cell killing (figure 4B). Cumulatively, these data show that progesterone treatment, working through PR, leads to downregulation of MHC class I expression on PR-expressing tumor cells, which aids in protection from immune cell-mediated death.

PR expression in tumor cells shields from immune-mediated killing. (A) Splenocytes were isolated from OT-I C57BL/6 mice and cultured with E0771-OVA-vec or E0771-mPR for 3 days. The media were supplemented with 20 nM progesterone or control. After 72 hours, non-adherent cells were removed, remaining live cells were lysed, and luciferase activity was measured. All conditions included n=6 replicates, with error bars representing the SD. Statistical significance between groups at each ratio was determined using multiple t-tests with Bonferroni correction; *p≤0.05. (B) Assay performed as in (A), with E0771-H-2Kb expressing E0771 variants. Statistical significance between groups at each ratio was determined using multiple t-tests with Bonferroni correction. All comparisons were NS=not significant, with the exception of two individual data points (these data points are marked with a star as the data point marker, instead of a circle, square, or triangle); *p≤0.05. PR, progesterone receptor.
PR-dependent protection from immune-mediated death requires CD8+ T cells
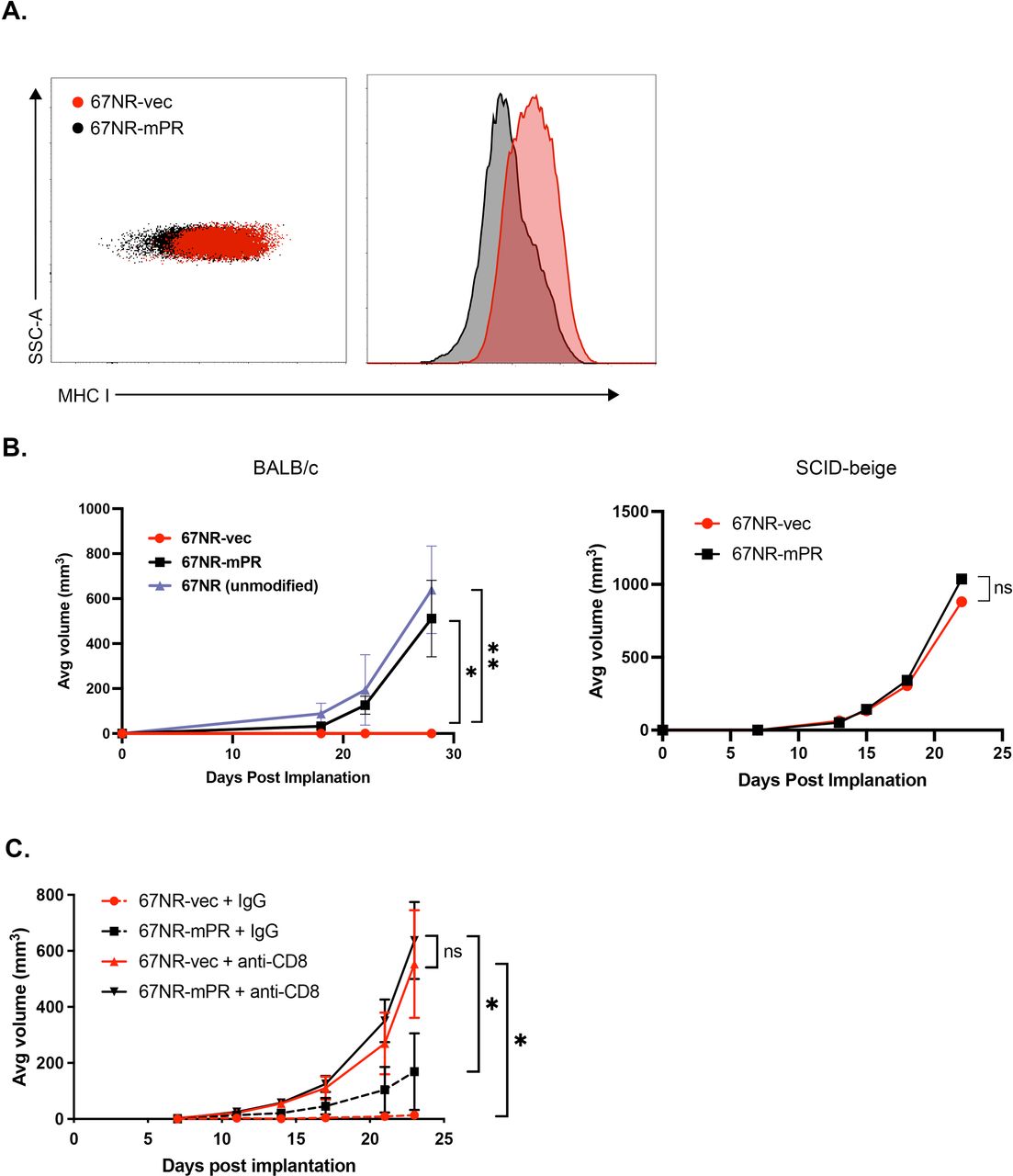
Using an additional mouse tumor cell line, 67NR47–49 (also engineered to express mPR or a matching empty vector, non-PR-expressing construct; online supplemental figure 5), we observed that exogenous expression of PR led to decreased baseline MHC class I expression compared with 67NR-vec cells (figure 5A). To determine whether this downregulation in MHC class I results in tumor protection, we injected 67NR-mPR, 67NR-vec, or unmodified parental 67NR cell lines into the mammary fat pads of immunocompetent BALB/c mice. Following orthotopic implantation into syngeneic BALB/c mice, we found that the stable expression of hygromycin or GFP on a lentiviral vector backbone triggered complete rejection of these tumors in BALB/c mice, but the expression of PR permitted tumor growth comparable to parental (non-xenoantigen-expressing) 67NR cells. Critically, growth was identical between control or PR-expressing 67NR lines in SCID-beige mice (defective natural killer cells, and deficient for T and B cells), suggesting that rejection was immune-mediated (figure 5B). To determine whether cytotoxic CD8+T cells were responsible for 67NR-vec tumor clearance, 67NR-mPR or 67NR-vec cells were injected into immune-competent mice that received anti-CD8 antibodies (or isotype control) to systematically deplete CD8+T cells (figure 5C and online supplemental figure 6). Interestingly, depletion of CD8+T cells confers similar tumor growth between 67NR-mPR and 67NR-vec. These findings demonstrate that PR signaling in 67NR-mPR cells plays a crucial role in downregulating MHC class I expression, thereby inhibiting CD8+T cell recognition and cytotoxicity, allowing for immune evasion and tumor growth.

PR-induced MHC I downregulation facilitates immune evasion from CD8+T cells. (A) Flow cytometric analysis of MHC I expression on untreated 67NR tumor cells stably expressing either empty vector (67NR-vec) or mouse PR (67NR-mPR). (B) 67NR-vec, 67NR-mPR, or unmodified 67NR parental cells were injected into the mammary fat pads of immunocompetent BALB/c mice (left panel) or immunocompromised SCID-beige mice (right panel). (C) 67NR-vec or 67NR-mPR cells were injected into BALB/c mice and treated with either anti-CD8 monoclonal antibodies (mAb) or IgG control biweekly for 4 weeks. Error bars represent STD. Differences in tumor growth over time among the groups were analyzed using linear mixed models for repeated measure data using SAS procedure GLIMMIX AR(1) covariance structure (see Methods); *p≤0.05, **p≤0.01. MHC, major histocompatibility complex; ns, not significant; PR, progesterone receptor; STD, standard deviation.
Effective inhibition of PR+ tumor growth by antiprogestins is immune-mediated
To determine if inhibition of PR activity in our mouse tumor models would reverse PR+tumor growth, we treated 67NR-PR-tumor bearing mice (BALB/c) with two different antiprogestins: mifepristone (RU486) and onapristone (or placebo control). Mifepristone is the gold standard antiprogestin, currently used clinically as an abortifacient.50 While very effective at blocking PR action, it also has effects on the GR. We also used onapristone, a newly developed antiprogestin that was recently evaluated in phase II clinical trials for endometrial cancer, and in window of opportunity trials for BC.51 52 Onapristone demonstrates superior suppression of PR/progesterone signaling at a molecular level, and thus may allow for a more favorable therapeutic suppression of PR signaling with reduced off-target effects.53–58 When mice were treated with either antiprogestin, 67NR-PR tumor growth was completely abrogated (figure 6—left). We repeated these experiments in SCID-beige mice to determine the contribution of the immune system to this phenotype, and observed that antiprogestins could only block∼50% tumor growth in mice lacking an immune system (figure 6—right). These data suggest that antiprogestins prevent PR+tumor growth, at least in part, through an immune-mediated mechanism.

Inhibition of PR+tumor growth by antiprogestins is immune-mediated. 30 mg mifepristone, onapristone, or placebo pellets were implanted subcutaneously into the neck of immunocompetent BALB/c mice (left panel) or immunocompromised SCID-beige mice (right panel). 7 days following pellet insertion, 67NR-mPR cells were injected into the mammary fat pads of recipient mice, and tumor growth was measured over time. Error bars represent STD. Differences in tumor growth over time among the groups were analyzed using linear mixed models for repeated measure data using SAS procedure GLIMMIX AR(1) covariance structure (see Methods); ***p≤0.001. PR, progesterone receptor; STD, standard deviation.
APP genes are lower in patients with PR+ BC
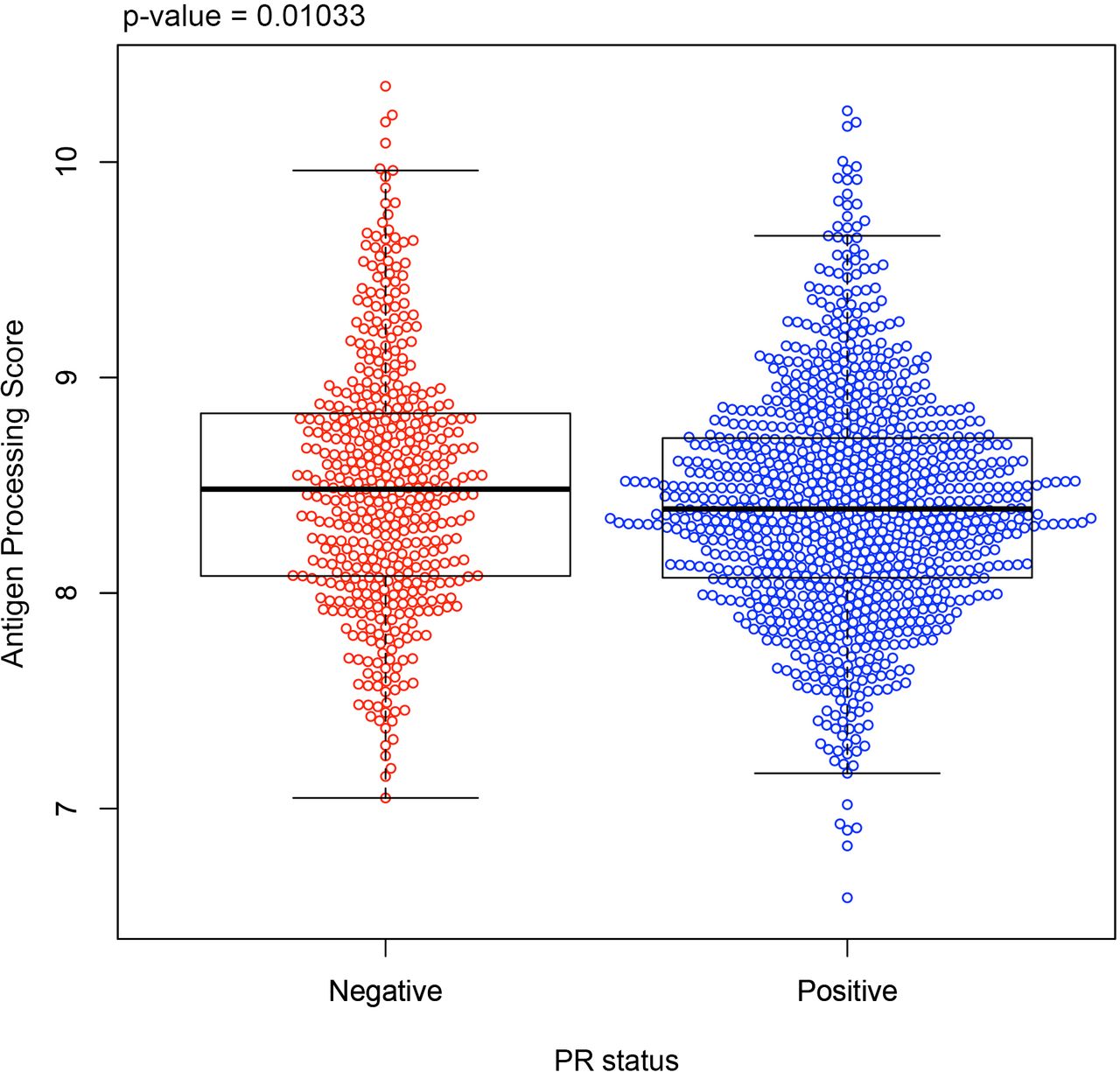
To evaluate if human BCs show similar APP/MHC class I downregulation correlated with PR expression, we examined APP gene expression in 1,460 human BCs from the METABRIC data set. Analyzed tumor samples were all positive for ER expression, and were further stratified based on expression of PR. When combining all APP genes together into a composite gene score (HLA-A, HLA-B, HLA-C, NLRC5, B2M, TAP1, TAP2, TAPBP, PSMB8, and PSMB9), tumors that were ER+/PR+ (positive for both ER and PR) had a lower APP score (figure 7) compared with ER+/PR-negative (positive for ER, negative for PR) tumors. Note that while patient data showed modest changes in APP/MHC class I, these changes were widespread across many patients, who may have different levels of circulating hormones or different menopause status. Collectively, these data show that PR plays an important role in downregulating APP machinery and MHC class I in human HR+BC.

APP gene expression is lower in PR-positive human breast tumors. The antigen processing and presentation (APP) gene score (see Methods) is shown for all ER-positive tumors in the METABRIC database, sorted by PR positivity; PR-negative (red) and PR-positive (blue). APP genes included in the composite gene score: HLA-A, HLA-B, HLA-C, NLRC5, B2M, TAP1, TAP2, TAPBP, PSMB8, and PSMB9). The statistical test was carried out using a Wilcoxon rank-sum test. ER, estrogen receptor; METABRIC, Molecular Taxonomy of Breast Cancer International Consortium; PR, progesterone receptor.
Surface MHC class I expression is lower in patients with PR+BC
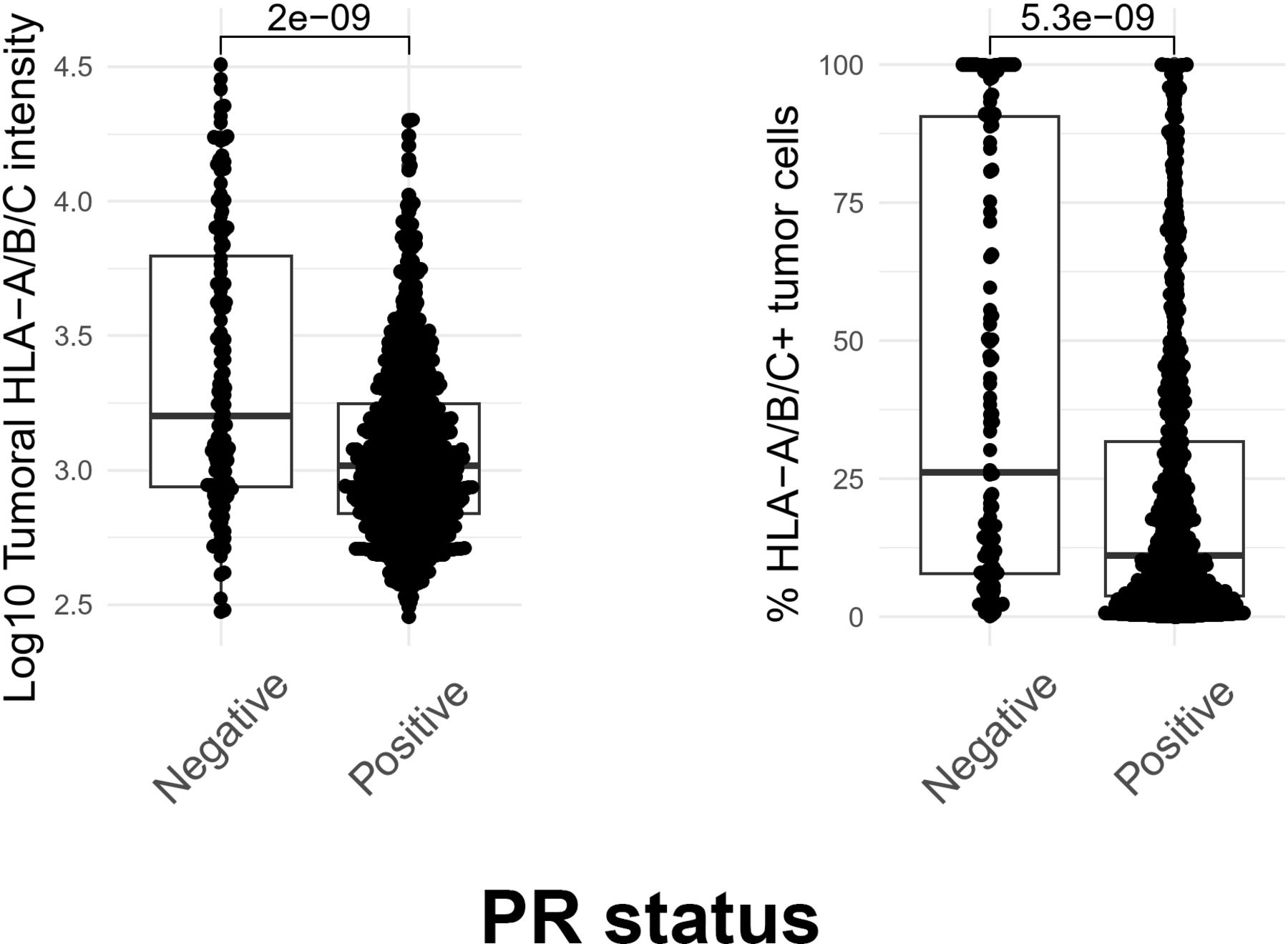
Building on our finding of APP downregulation in human BCs, we explored the relationship between HLA (surface MHC expression) and PR status. To determine if there is a correlation between HLA expression and PR positivity at the protein level, we examined data from the Carolina Breast Cancer Study (CBCS). The CBCS is a population-based study conducted in North Carolina that began in 1993. Briefly, cases of invasive BC between age 20 and 74 years were identified using rapid case ascertainment in cooperation with the NC Central Cancer Registry, with black and young cases (age 20–49 years) oversampled using randomized recruitment. Study details and sampling schemes have been described previously.39 For this study, we selected 992 patients with HR+/HER2− BC who are classified as PR positive (>10% tumor cells expressing PR) or PR negative (<1% tumor cells expressing PR), all ER positive. We found that human BCs expressing both ER and PR had lower pan-HLA levels (figure 8—left) and a lower percentage of HLA+ (figure 8—right) cells when compared with ER+/PR− tumors (positive for ER, negative for PR). These findings collectively suggest that human BCs from a large and diverse study population exhibit lower MHC expression when PR is expressed.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MHC Class I expression is lower in PR-positive human breast tumors. Tumor-specific MHC-I (HLA-A/B/C) expression was measured using immunofluorescent assays staining for CK and MHC-I (HLA-A/B/C) on formalin-fixed paraffin-embedded tissue sections from patients in the CBCS cohort. To evaluate the association between PR and MHC-I expression, we performed a two-sample t-test comparing log10 normalized MHC-I fluorescent intensity (left) and the percentage of MHC-I positive tumor cells (right) between PR positive and negative tumors (all ER positive), representing 992 tumors. P values are reported above each comparison. CBCS, The Carolina Breast Cancer Study; ER, estrogen receptor; MHC, major histocompatibility complex; PR, progesterone receptor.
Discussion
We reveal a novel mechanism by which progesterone signaling through PR promotes growth in PR+tumors by modulating the APP/MHC class I machinery, aiding in evasion from immune-mediated clearance. We demonstrate that progesterone alone regulates HLA and APP machinery, but this progesterone-dependent downregulation of MHC class I/APP genes is more pronounced when combined with IFNγ treatment. We show that knockdown of PR in T47D BC cells increased APP gene levels/MHC class I expression, and progesterone treatment decreased surface MHC class I only in cells with robust PR expression. Furthermore, in vitro immune killing assays reveal that E0771-OVA-mPR, when exposed to progesterone, are protected from immune cell-mediated cytotoxicity. Moreover, we showed that APP gene expression and MHC Class I expression were downregulated in PR+patients, as compared with PR− patients, in two large clinical data sets. Together, our data suggest that progesterone signaling through PR leads to transcriptional downregulation of APP genes and MHC class I expression, leading to evasion from immune cell killing and subsequent tumor growth.
Clinical aspect of PR biology
Clinically, PR is used as a surrogate marker for functional ER and is used to predict response to endocrine therapy.59 This is because canonical ER signaling induces expression of PR in most BC cells. Therefore, high PR expression, an indicator of robust ER signaling, is a sign that women will respond well to antiestrogen or anti-ER-based endocrine therapies. While robust ER/PR expression is associated with a better prognosis, we postulate that the tumor-promoting effects of PR described here may be stage dependent. PR-specific downregulation of APP/MHC class I may play a key role in early tumor formation but also, importantly, metastatic seeding/colonization and tumor outgrowth by impairing immune surveillance. Moreover, there have been reports suggesting that PR is important in early lesion development60–62 and is also implicated in early dissemination of tumor cells during the initial stages of tumor formation.63 Progesterone, acting through PR, plays a crucial role in the differentiation and proliferation of mammary gland tissue. This occurs via paracrine signaling, where PR-positive cells produce RANKL, which then acts on its receptor RANK, expressed on PR-negative cells. Notably, in both normal mammary tissue and BC, RANKL expression has been shown to tightly correlate with serum progesterone levels.64 This strong association underscores a potential link between progesterone’s effects on mammary gland development and BC formation, potentially mediated by RANKL-dependent proliferative mechanisms.64 65 Furthermore, Graham et al established a model to better recapitulate human ER/PR heterogeneity and PR-mediated paracrine signaling by using organoids isolated and generated from reduction mammoplasties. Their findings demonstrated that progesterone induced the proliferation of progenitor stem cells (CK5+MUC1+p63−) in PR-negative cells, providing compelling evidence that progesterone exerts its effects indirectly through paracrine signaling from PR-positive cells.66 67 This mechanistic insight further reinforces the connection between PR/progesterone signaling and BC incidence, as dysregulation of this pathway may drive tumor initiation and progression.
Finally, to determine the role of the PR function, it is important to control for factors such as tumor stage and menopause status. For BC to be categorized as ER/PR positive, at least 1% of cells must express either receptor. However, a tumor with 1% receptor positivity differs significantly from one with 90% positivity.68 69 This variability should be controlled for in clinical studies when investigating the role of PR/progesterone in tumor development and treatment outcomes. Advances in spatial technology will provide a better understanding of how PR expression within the same tumor influences gene expression and the surrounding immune cell microenvironment.
Immunosuppressive effects of progesterone
The field of pregnancy research, specifically at the maternal–fetal interface, has shed light on the important role sex-steroidal hormones play in modulating various components of the immune system. Pregnancy is an interesting time to study the immune system, given high concentrations of progesterone and estrogen that last the entire pregnancy and facilitate the rewiring of the immune system, particularly at the maternal–fetal interface. During a healthy pregnancy, a critical balance between immune tolerance and robust immune defense against pathogens must be established. Without the ability to establish immune tolerance, the mother’s body may reject the semiallogeneic fetus. Conversely, if the mother’s immune response is too weak, she becomes susceptible to viral infections, which is well-documented as pregnancy is often considered an immunosuppressive state with an increased risk of viral infections.70–74 In the initial stages of an immune response to bacterial or viral infections, neutrophils and natural killer cells release type I and II IFNs, creating inflammatory conditions that activate the immune system. However, excessive inflammation at the maternal–fetal interface can induce miscarriage. Our hypothesis suggests that cell-intrinsic steroid receptor signaling, specifically through PR, may mitigate this risk by modulating the response to local inflammation at the maternal–fetal interface, thereby shielding the fetus against the detrimental effects of IFN signaling. This concept aligns with our laboratory’s publications detailing how PR/progesterone signaling can dampen the impact of IFNs.6–8 The presence of high endogenous levels of progesterone and estrogen during pregnancy helps protect maternal–fetal interface cells from inflammation, allowing the immune system to effectively combat pathogens without compromising the fetus. We propose that ER/estrogen and PR/progesterone play a pivotal role in facilitating a balanced immune response that prevents fetal rejection while still controlling infection, primarily by modulating the effects of type I and II IFNs. During pregnancy, immune cell functionality is notably different. For instance, neutrophils isolated from blood during the first trimester are less responsive to IFNγ and exhibit dysfunctional phagocytic capabilities and nitric oxide production.75–77 Moreover, maternal CD8 T cells are primed against fetal antigens but have dysfunctional effector functions78–80 and the presence of progesterone increases the proportion of CD4+CD25+Foxp3+ Treg cells.81 It remains unclear which of the immunosuppressive mechanisms that occur during pregnancy are hijacked to allow the formation of hormone-driven cancers.
Extensive research on the immunosuppressive effects of sex-steroidal hormones highlights striking parallels with the immunosuppressive phase of postpartum mammary gland involution, which may inadvertently increase BC risk. Postpartum mammary gland involution, a physiologically normal process involving tissue remodeling, inflammation, and extracellular matrix deposition, mirrors pathways observed in wound healing and has been implicated in the increased risk and poor prognosis of BC diagnosed within 5–10 years after childbirth.82 Specifically, involution-associated processes, including lymphangiogenesis, immunosuppressive macrophage activity, and stromal remodeling, have been shown to promote metastasis in both preclinical and clinical studies.82–84 These findings suggest that the hormonal and immunological adaptations essential during pregnancy may inadvertently provide a permissive niche for the development and dissemination of hormone-driven cancers.
In this study, we demonstrate that one of the mechanisms progesterone uses to induce immune suppression in PR+tumors is through the downregulation of APP/MHC class I machinery. It is well established that various types of tumors, including head and neck,85 colorectal,86 pancreatic,87 melanoma,80 88 89 and lung cancer90 use multiple mechanisms to modulate APP/MHC class I expression, which has been extensively documented in the literature. However, the role of progesterone in regulating immune evasion in the context of HR+BC remains unknown.
Steroid HRs and control of MHC class I
Previous research from our laboratory demonstrated that PR/progesterone signaling suppresses type I–II IFNs by inhibiting STAT1 phosphorylation7 and promotes STAT2 degradation6 resulting in less ISG transcription.8 We are not the first to publish on the link between steroid receptors and APP/MHC class I28 29; however, to the best of our knowledge, this is the first time PR/progesterone has been implicated in the regulation of MHC class I. A previous study has shown that tumor-intrinsic signaling of GR in pancreatic ductal adenocarcinoma leads to the downregulation of APP/MHC class I. This effect was reversed through genetic knockdown of GR or pharmacological inhibition using a GR antagonist (mifepristone). Furthermore, combination therapy with mifepristone and checkpoint blockade resulted in robust CD8 T cell-dependent tumor control.87 Additionally, dexamethasone, a GR agonist, is given for treatment of autoimmune/inflammatory diseases such as rheumatoid arthritis, lupus, asthma, and inflammatory bowel disease (as reviewed in91–94). Moreover, another group conducted a window of opportunity study to understand the effect of endocrine therapy in patients with HR+BC. Women were treated with tamoxifen or letrozole based on their menopausal status. The study found that after endocrine therapy, TIL signatures increased, and B2M levels rose as a surrogate marker for HLA. However, it remains unclear whether the modulation in TIL infiltrates or the increase in HLA was due to decreased ER signaling or decreased PR signaling, given both were reduced after endocrine therapy.28 Furthermore, when examining TILs across different BC subtypes, the HR+/HER2− subtype exhibits the lowest percentage of TILs compared with other subtypes.95–97 Additionally, studies comparing HLA expression relative across BC subsets have shown that HR+/HER2− cancers have lower HLA expression relative to the other types.98 99 Our data, along with previous reports, indicate that these effects may be reversible. For instance, treating MCF7 cells with fulvestrant, a selective ER degrader, resulted in an increase in total MHC class I expression.29 Several clinical trials have explored the use of pembrolizumab or nivolumab, programmed cell death protein-1 inhibitors, in combination with various strategies to enhance antitumor immunity against HR+BC, including radiation, chemotherapy and adjuvant endocrine therapy.22 24 26 Based on these novel non-canonical mechanisms of immune regulation by HRs we hypothesize that administering neoadjuvant endocrine therapy followed by pembrolizumab may improve patient outcomes. It appears that the pleiotropic role of steroid receptors varies depending on whether it is tumor or immune intrinsic signaling. However, they generally appear to regulate the immune system in various ways. The expression of APP/MHC class I across tumors may be one of the mechanisms that tumors broadly use to impact immunosurveillance. We propose that targeting both ER and PR may restore immunosurveillance, potentially enhancing the effectiveness of immunotherapies. While our data demonstrate modest decreases in MHC class I expression, enhanced protection from immune cell-mediated killing, and impaired antigen processing, it is important to note that we did not assess the quality of antigens presented by MHC class I. This limitation leaves open the possibility that progesterone signaling might also affect antigen quality, potentially leading to weak T cell activation, which warrants further investigation.
Data availability statement
Data sharing not applicable as no data sets generated and/or analyzed for this study.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the University of North Carolina Institutional Review Board (IRB 92-0410 and IRB 21-0956) in accordance with US Common Rule. All study participants provided written informed consent prior to study entry. This study complied with relevant ethical regulations, including the Declaration of Helsinki.
References
Footnotes
X @balkolab, @HaganlabKUMC
JCT and HIS contributed equally.
Contributors Conception and design: JCT, HIS, HM, MAM, ZCH, CRH. Writing: JCT, HIS, ZCH, CRH. Critical review and editing: all authors contributed to the critical review and editing of the manuscript. Data acquisition: all authors contributed to data acquisition in the manuscript. Guarantor author: CRH.
Funding This work was supported by: NCI Cancer Center Support Grant P30 CA168524 (CRH), funds from the University of Kansas School of Medicine (CRH), NCI R21CA274044 (CRH and ZCH), DOD BC201209 W81XWH-21-1-0349 (CRH), ACS RSG-23-1152337-01-IBCD (CRH), Breast Cancer Alliance AWD10000196 (CRH), Context Therapeutics (CRH), NCI F30CA271796 (LRW), Susan G Komen ASPIRE ASP241267015 (JCT), Susan G Komen ASPIRE ASP231051198 (HIS). This work made use of the University of Kansas Medical Center Flow Cytometry Core Laboratory, which was started with funds from the NIH/NIGMS COBRE grant P30 GM103326 and is currently partially supported by the NIH/NCI Cancer Center grant P30 CA168524. Additionally, this work was supported by an Institutional Development Award (IDeA) from the NIGMS under grant number P20GM103418. Investigators affiliated with the Carolina Breast Cancer Study (MAT and CP) were supported by the National Cancer Institute Specialized Program of Research Excellence (SPORE) in Breast Cancer (NIH/NCI P50-CA058223).
Competing interests CRH received funds from Context Therapeutics (former developer of onapristone). JMB receives research support from Genentech/Roche and Incyte Corporation, has received advisory board payments from AstraZeneca, Eli Lilly, and Mallinckrodt, and is an inventor on patents regarding immunotherapy targets and biomarkers in cancer. The remaining authors have nothing to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.