Article Text

Abstract
Background The pleiotropic roles of tumor-associated macrophages (TAMs) render them attractive targets in antitumor drug development. CD47/SIRPα (signal regulatory protein alpha) and CD24/Siglec-10 (sialic acid-binding immunoglobulin-like lectin 10) signaling pathways have been found to suppress macrophage phagocytosis of malignant cells. Systemic blockade of CD47/SIRPα has shown severe side effects. Intratumoral delivery of a CD47 inhibitor by oncolytic viruses (OVs) may circumvent this hurdle.
Methods To identify the characteristics of recombinant adenovirus (AdV)-SIRPα/Siglec-10, we conducted CCK8 assay, quantitative PCR, western blot, competitive binding assay, in vitro cytotoxicity assay, ELISA and phagocytosis assay. We investigated the antitumor immune responses of AdV-SIRPα/Siglec-10 using flow cytometry, various tumor-bearing mouse models, humanized tumor-bearing mouse models, immune cell depletion, RNA sequencing, and in vitro T cell activation assay.
Results Here, we developed a novel AdV encoding a fusion protein composed of the extracellular domains of murine or human SIRPα and Siglec-10 (SIRPα/Siglec-10), termed AdV-mSS or AdV-huSS. The SIRPα/Siglec-10 was effectively secreted by cells infected with AdV-mSS and functioned as a dual blocker of CD47 and CD24, thereby significantly enhancing macrophage phagocytosis. In a series of tumor models, including subcutaneous and ascitic H22 hepatocellular carcinoma (HCC), subcutaneous Hepa1-6 HCC, MC38 colorectal carcinoma, and Lewis lung carcinoma, AdV-mSS treatment markedly enhanced antitumor efficacy. Mechanistically, AdV-mSS reprogrammed TAMs toward an antitumor phenotype and enhanced the expression of major histocompatibility complex (MHC)-I/II, promoting CD8+T cell proliferation and activation. Depletion of either macrophages or CD8+T cells abrogated the antitumor efficacy of AdV-mSS. Similarly, in a humanized LM3 HCC mouse model, AdV-huSS significantly inhibited tumor growth and prolonged survival.
Conclusions Dual SIRPα/Siglec-10 inhibitor delivered intratumorally by AdV not only reinvigorated the TAM-CD8+T cell axis but also potentially reduced the risk of off-target effects. Further investigation of AdV-huSS in patients with cancer is warranted in the near future.
- Oncolytic virus
- Immune Checkpoint Inhibitor
- Macrophage
Data availability statement
Data are available upon reasonable request. The RNA-seq data used in this study have been deposited at the Gene Expression Omnibus under accession codes (SuperSeries GEO accession, GSE275905). The remaining data generated or analyzed during this study are available within the article and Supplementary Information. Source data are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The systemic administration of CD47/SIRPα (signal regulatory protein alpha)-based antitumor therapeutics has shown significant efficacy in hematologic malignancies and various solid tumors. However, its use is often limited by associated toxicities, such as hemolysis.
WHAT THIS STUDY ADDS
We developed recombinant adenoviruses (AdVs) for the first time that encode bispecific fusion proteins of SIRPα/Siglec-10 (sialic acid-binding immunoglobulin-like lectin 10, AdV-SS) to block both CD47/SIRPα and CD24/Siglec-10 checkpoints. Intratumoral administration of AdV-SS not only revitalizes the antitumor activity of tumor-associated macrophages (TAMs) but also induces TAM-CD8+T cell axis-mediated antitumor effects, while minimizing the risk of off-target effects.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study provides a potentially safe and effective antitumor candidate for clinical cancer therapy.
Introduction
Cancer immunotherapy is revolutionizing the treatment of several types of cancer.1 However, the majority of patients with solid tumors do not respond to current immunotherapy agents.2 One of the factors responsible for therapeutic resistance is tumor-associated macrophages (TAMs).3 Macrophages, a subset of terminally differentiated monocytic phagocytes, have emerged as promising targets in the field of tumor immunotherapy due to their phagocytic capacity, crucial roles in combating tumor cells, and key involvement in the crosstalk between the adaptive and innate immune systems, including antigen presentation.4 5 TAMs constitute the majority of infiltrating leukocytes within the tumor microenvironment (TME) of solid tumors.6 However, these TAMs are often reprogrammed toward the alternatively activated M2 phenotype and play a tumor-promoting role by suppressing antitumor immune responses, promoting angiogenesis, providing an immunosuppressive environment, and facilitating immune escape of tumor cells.3 4 6 Consequently, efforts are underway to reprogram TAMs from an immunosuppressive to an immunostimulatory phenotype.7
Tumor cells can evade TAM clearance by overexpressing antiphagocytic surface proteins known as “don’t eat me” signals, such as programmed cell death ligand 1,8 beta-2 microglobulin subunit major histocompatibility class I complex,9 CD4710 and CD24.11 Blocking these “don’t eat me” signals with antibodies has demonstrated therapeutic potential across various cancers.8 9 11 12
CD47, a receptor for members of the platelet response protein family, is present on the surface of many cells throughout the body on the surface of many cells in the body.12 Nearly all types of cancer cells express high levels of CD47 on their surfaces.12 CD47 binds to its receptor, signal regulatory protein alpha (SIRPα), on TAMs, thereby impairing phagocytosis.13 Blocking CD47 signaling with anti-CD47 antibody14 or fusion protein15 has been reported to stimulate macrophages to eliminate tumor cells. However, erythrocytes also highly express CD47 on their surface. Systemic blockade of CD47 often induces hemolysis, prompting numerous efforts to minimize the off-target effects of CD47 inhibition.12–15 Besides CD47, the tumor cell surface protein CD24 interacts with sialic acid-binding immunoglobulin-like lectin 10 (Siglec-10) on TAMs to transmit immunosuppressive signals.16 Barkal et al found that dual blockade of CD24 and CD47 signals further enhances phagocytosis of human tumors expressing CD24, resulting in the inhibition of tumor growth and prolonged survival time.11 Therefore, simultaneous targeting of CD47 and CD24 represents a promising therapeutic approach to enhance the antitumor capabilities of TAMs in the TME.
Oncolytic viruses (OVs) offer a versatile therapeutic platform when combined with other regimens in cancer immunotherapy.17–22 OVs can be delivered locally to selectively infect and replicate in cancer cells and cancer-associated stromal cells. OVs can also be genetically manipulated to express transgenes that coordinate antitumor forces in the TME, eradicating malignant cells.17–19 Crucially, OVs not only trigger direct lytic activities but also activate local immune cell-mediated antitumor responses, thereby augmenting the efficacy of delivered payloads.20 21 OVs also have a tolerable safety profile and have undergone clinical trials.22 23
In the current study, we generated a novel recombinant oncolytic adenovirus (AdV) encoding fusion proteins that specifically bind to both CD47 and CD24 (AdV-SS). We investigated its capacity to locally block both CD47/SIRPα and CD24/Siglec-10 “don’t eat me” signals in TAMs, its antitumor efficacy in a series of tumor models, and the underlying antitumor mechanisms. Our research provides a potential biotherapeutic strategy for tumor treatment.
Materials and methods
Cell culture
Murine bone marrow-derived macrophages (BMDMs) and T cells were collected as described previously in a study by Barkal et al.9 The femurs and tibias were isolated from 8–10 weeks old C57BL/6J and BALB/c mice. Bone marrow was flushed out, passed through 70 µm cell strainers, and resuspended in ammonium chloride potassium bicarbonate (ACK) lysis buffer at room temperature (RT) for 5 min to remove blood cells. Remaining cells were incubated at 2.5×106 cells/mL in complete Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 10 ng/mL mouse macrophage colony-stimulating factor (M-CSF) for 7 days.
To obtain human monocyte-derived macrophages (MDMs), human PBMCs isolated from whole blood by Procell were thawed and seeded in 35 mm plates at 2.5×106 cells/mL. After 90 min, non-adherent cells were aspirated, and adherent monocytes were incubated in complete Roswell Park Memorial Institute (RPMI) 1640-GlutaMAX (Gibco) with 25 ng/mL human M-CSF for 7 days. MDMs were collected when they exhibited numerous cytoplasmic granules and a slightly elongated spindle shape.
Naïve T cells or OT-I T cells were isolated from the spleens of naive mice or OT-I mice and seeded in 60 mm2 plates at 1×106 cells/mL in complete RPMI 1640 (Gibco) with CD3ε/CD28 antibodies (6-8900-050, IBA Life Sciences) and 10 ng/mL interleukin-2 for 5 days.
The mouse hepatocellular carcinoma lines H22 and Hepa 1–6 were purchased from the China Center for Type Culture Collection (Wuhan, China). The mouse colon carcinoma cell line MC38 was purchased from the National Cancer Institute (USA). The mouse Lewis lung carcinoma (LLC) cell line, human hepatocellular carcinoma cell line (LM3), and 293T cells were purchased from the American Type Culture Collection (USA). H22-CAR, MC38-CAR, LLC-CAR, H22-CAR-Luc, MC38-CAR-Luc, H22-OVA, H22-YFP, MC38-YFP, LM3-YFP, and H22-YFP-CAR cell lines were established by transducing wild-type cell lines with lentiviruses and selecting with 2 µg/mL or 4 µg/mL puromycin according to the manufacturer’s instructions.
Adenovirus construction
All lentiviruses used in this study were purchased from OBiO Tech (Wuhan, China). Prior to investigations, mycoplasma infection was ruled out in all cell lines. Cell lines were cultured in the appropriate medium (Gibco) supplemented with 10% fetal bovine serum (10099158, Gibco), 100 U/mL penicillin, and 100 µg/mL streptomycin (15140163, Gibco), in accordance with Cellbank recommendations. All cells were maintained at 37 °C under an atmosphere containing 5% CO2.
The recombinant AdV was constructed as described in our previous study.24 The adenoviral shuttle plasmid pENTR vectors, encoding viral E1A along with the extracellular domains of SIRPα and Siglec-G, were obtained from Genscript, as depicted in figure 1A. Recombinant adenoviral vectors expressing fusion proteins were generated via homologous recombination between the shuttle plasmid and the AdV type 5 backbone pAd/PL-DEST (Invitrogen), followed by digestion with the restriction enzyme PacI. Following transfection into 293T cells, the viruses were identified, amplified, and purified.

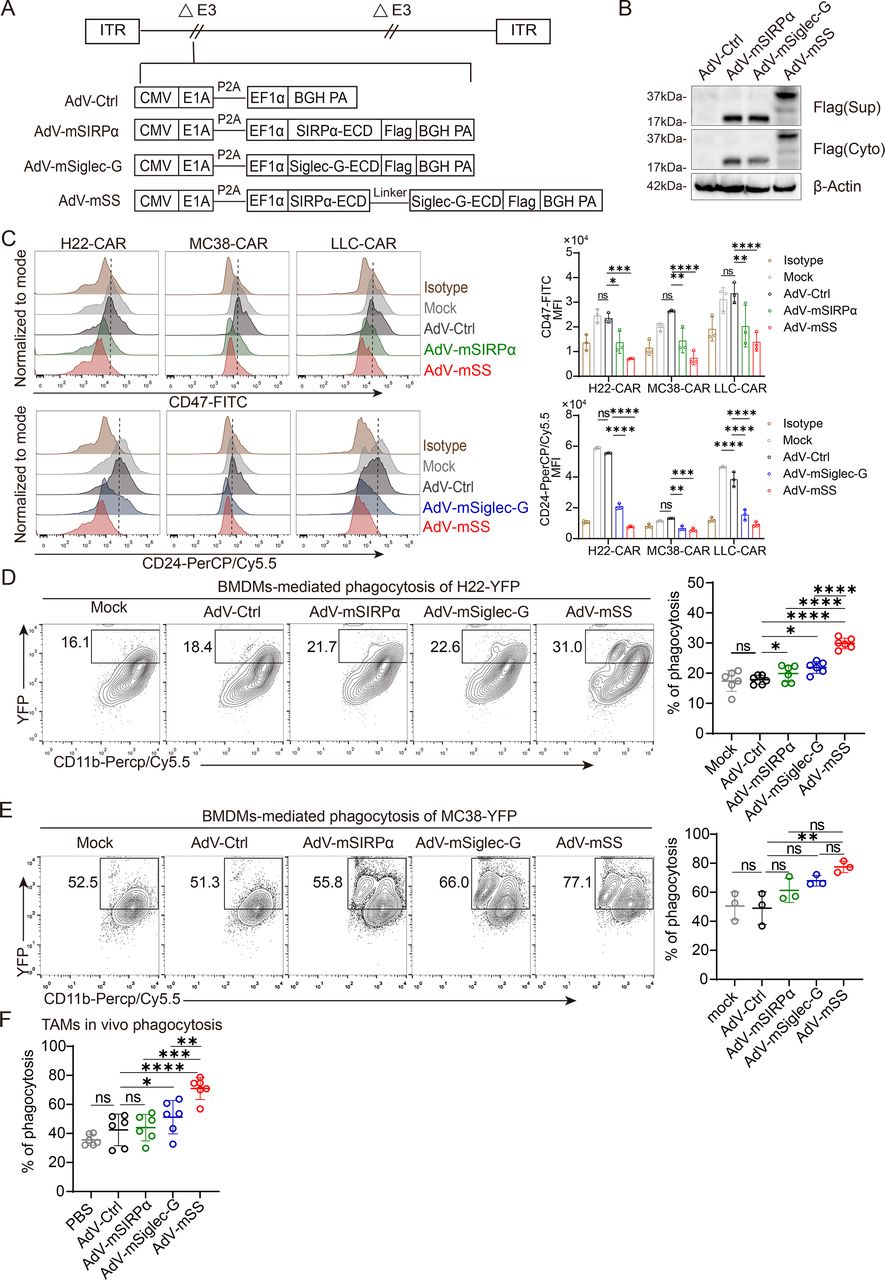
Dual blocker mSIRPα/Siglec-G expressed by recombinant AdV-mSS enhances phagocytosis of macrophages. (A) Schematic structure of recombinant oncolytic adenovirus used in this study. (B) Western blot detection of Flag-labeled fusion protein expression in OV-infected 293T cells. Culture supernatants and cell lysates were collected to identify soluble mSIRPα, soluble mSiglec-G or soluble mSIRPα/Siglec-G protein levels. n=3 independent experiments. (C) The binding ability of fusion proteins to multiple mouse tumor cells was detected by competitive flow cytometry. FITC-conjugated anti-mouse CD47 antibody or PerCP-Cy5.5-conjugated anti-mouse CD24 antibody competes with fusion proteins purified from the supernatants of OV-infected 293T cells for binding to tumor cells. n=3 independent experiments. (D) Quantification of in vitro phagocytosis against H22-YFP cells by mouse BMDMs with the supernatants from OV-infected 293T cells. The percentage of BMDMs phagocytosis against H22-YFP cells (CD11b+YFP+ cells) was calculated by flow cytometry. n=6 independent experiments. (E) Quantification of in vitro phagocytosis against MC38-YFP cells by mouse BMDMs with the supernatants from OV-infected 293T cells. Percent of BMDMs phagocytosis against MC38-YFP cells (CD11b+YFP+ cells) were calculated by flow cytometry. n=3 independent experiments. (F) Quantification of in vivo phagocytosis against H22-YFP-CAR cells by TAMs in H22-YFP-CAR ascites model. The percentage of TAMs phagocytosis against H22-YFP-CAR cells (CD45+CD11b+F4/80+YFP+ cells) were calculated by flow cytometry. n=6 independent experiments. All data are the mean±SD. Statistical analyses were determined using two-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons test (C) and one-way ANOVA followed by Bonferroni multiple comparisons test (D–F) (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-mSS, AdV-mSIRPα/Siglec-G; BMDMs, bone marrow-derived macrophages; BGH PA: bovine growth hormone polyadenylation; CAR, coxsackievirus and adenovirus receptor; CMV: cytomegalovirus; ECD, extracellular domain; EF1α: elongation factor 1 alpha; FITC: fluorescein Isothiocyanate; LLC, Lewis lung carcinoma; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OV, oncolytic virus; TAMs, tumor-associated macrophages; YFP: yellow fluorescent protein.
For viral stock production, the titer of infectious particles was quantified by detecting viral hexon protein production in infected cells through immunohistochemistry. 293T cells were seeded into a 24-well plate (2.5×105 cells/well) and infected with serial dilutions of AdV. After 48 hours, the cells were fixed and treated with a hexon protein-specific antibody (ab2596, Abcam), an horseradish peroxidase (HRP)-conjugated antibody (31430, Invitrogen), and developed with 3,3'-Diaminobenzidine (DAB) Substrate (PK10005, Proteintech). The virus titer was calculated by counting the number of stained cells in a given area under a 20× objective, each of which corresponds to a single infectious unit (IFU).
The virus titer for each well was calculated as follows:

Tumor tissues were lysed with a homogenizer, and the supernatants were collected. Virus titers were calculated as stated above.
Viral oncolysis
The viral oncolysis was measured by CCK8 assay. Oncolytic AdVs were added to a 96-well plate seeded with tumor cells (1×104 cells/well) at the indicated multiplicity of infection (MOI). After 72 hours of infection, the cells were incubated for 4 hours with 20 µL of CCK8 solution (5 mg/mL; IM0280, Solarbio) added to each well. After discarding the supernatants, 150 µL of dimethyl sulfoxide (DMSO) was added to each well. The absorbance (A) was measured using a microplate reader (Spectra-Max M3, Molecular Devices, USA) at a wavelength of 450 nm. The cell viability was determined as follows:

Quantitative PCR
The AdV genome copy number was quantified by measuring hexon gene levels using quantitative PCR (qPCR).25 For the in vitro viral replication assay, tumor cells were seeded in 24-well plates (5×104 cells/well) and OVs were added at a MOI of 1. After incubation for 6, 12, 24, 48, 72, and 96 hours, 500 µL of cell lysis buffer (100 µg/mL proteinase K, 50 mM potassium chloride, 10 mM Tris, 0.5% Tween-20) was added per well, and cells were thoroughly pipetted. The cell lysate was heated at 56°C for 45 min and then at 100°C for 10 min to release viral genomic DNA for subsequent qPCR amplification. In vivo, tissues from mice, including the brain, cerebellum, heart, lung, liver, spleen, kidney, small intestine, and muscle, were collected 48 hours post-treatment. Genomic DNA was extracted using the TIANamp Genomic DNA Kit (DP304, TIANGEN) following the manufacturer’s instructions.
Stable cell line validation and messenger RNA expression of TAMs were assessed by real-time qPCR. Cells were resuspended and lysed in Trizol (Vazyme, Nanjing, China), and complementary DNA was synthesized using HiScript III RT SuperMix (R323, Vazyme) according to the manufacturer’s instructions. Standard qPCR was performed using a ViiA 7 Real-Time PCR System (Applied Biosystems) with SYBR qPCR Master Mix (Q511, Vazyme) and normalized to β-actin. All data were analyzed using QuantStudio Design and Analysis Software V.1.3 (Applied Biosystems). All primer sequences used in this study are listed in online supplemental table 1.
Supplemental material
Western blot
The expression of fusion proteins in supernatants and cell lysates was determined by western blotting. 48 hours after infection with OVs at a MOI of 5, supernatants and 293T cells were harvested and lysed using radio-immunoprecipitation assay (RIPA) buffer. Primary antibodies included the DYKDDDDK tag monoclonal antibody (MA1-91878, Invitrogen), Siglec-10 polyclonal antibody (PA5-55501, Invitrogen), and β-Actin monoclonal antibody (MA1-140, Invitrogen) as a loading control. Secondary antibodies were HRP-conjugated goat anti-mouse IgG (H+L) (A16068, Invitrogen) and goat anti-rabbit IgG (H+L) (31460, Invitrogen).
Competitive binding assay
The binding ability of fusion proteins was indirectly quantified using competitive flow cytometry as described in a previous study by Tian et al.26 Supernatants from 293T cells were collected 48 hours after infection with OVs. H22-CAR, MC38-CAR, LLC-CAR, or LM3 cells were suspended in supernatants from OVs-infected 293T cells at a concentration of 1×106 cells/mL and incubated for 2 hour at 37°C. Cells were washed three times with fluorescence-activated cell sorting (FACS) buffer (0.5% bovine serum albumin (BSA) in Phosphate-Buffered Saline(PBS)) and then stained with 10 µg/mL of Fluorescein Isothiocyanate (FITC)-conjugated anti-CD47 antibody and PerCP-Cy5.5-conjugated anti-CD24 antibody for 15 min at RT. Cells were then washed twice with FACS buffer. Binding of anti-CD47 monoclonal antibody (mAb) and anti-CD24 mAb to tumor cells was immediately detected by flow cytometry using BD FACS Calibur (BD Biosciences).
Phagocytosis assay
Phagocytosis analysis of macrophages was performed as previously described in a study by Gordon et al.8 For in vitro phagocytosis, mouse BMDMs or human MDMs were co-cultured with tumor cells expressing yellow fluorescent protein (YFP) in a U-bottom ultralow attachment 96-well plate at an Effector:Target ratio of 1:2 in supernatants from OVs-infected 293T cells for 4 hour at 37°C. All cells were harvested, washed with PBS to remove non-adherent cells, and incubated with cold Trypsin-EDTA at RT for 10 min to detach them. After phagocytosis, macrophages were stained with PerCP-Cy5.5-conjugated anti-CD11b antibody. Phagocytosis was quantified by measuring the percentage of CD11b+YFP+ cells using BD FACS Calibur (BD Biosciences).
For in vivo phagocytosis, 2×106 H22-YFP-CAR cells were injected into the peritoneal cavity of BALB/c mice to establish an HCC ascites model. When ascites formed, mice were randomly grouped and intraperitoneally (i.p.) injected with OVs or PBS (5×107 IFU per dose, every other day) for two doses. 48 hours after treatment, ascites cells were obtained by paracentesis from the peritoneal cavity of tumor-bearing mice. Phagocytosis of TAMs was quantified by measuring the percentage of CD45+CD11b+F4/80+YFP+ macrophages using CytoFLEX LX (Beckman Coulter). Data were analyzed using FlowJo V.10 (Treestar).
In vitro cytotoxicity assay
The in vitro cytotoxicity assay was based on firefly luciferase (Fluc) release, as described previously in a study by Corbett et al.27 Supernatants from 293T cells were collected 48 hours after infection with OVs. Subsequently, mouse BMDMs were co-cultured with tumor cells stably expressing Fluc in the presence or absence of naïve T cells at a ratio of 1:1:1 in supernatants from OVs-infected 293T cells for 48 hours at 37°C. Human MDMs were co-cultured with tumor cells stably expressing Fluc at a ratio of 1:1:1 in supernatants from OVs-infected 293T cells for 48 hours at 37°C. After incubation, cells were collected, centrifuged, and supernatants were separated. Remaining cells were washed once with precooled PBS to remove dead cells and lysed with adenosine triphosphate (ATP) lysis buffer. One mM D-luciferin (Promega) was added to the ATP cell lysate plated in a black 96-well plate at a 1:4 ratio (v/v) for immediate luciferase assays. Relative luminescent units were read in endpoint mode using the BioTek Synergy H4 hybrid microplate reader for 10 s.
Cytokine release assay
Tumor necrosis factor (TNF)-α release by mouse BMDMs or human MDMs was determined using the ELISA MAX Deluxe Set TNF-α (430904/430204, BioLegend). The assays were performed with 50 µL of supernatant separated from in vitro cytotoxicity assay stated above according to the supplier’s protocol. Interferon (IFN)-γ release by OT1 CD8+T cells was determined using ELISA MAX Deluxe Set mouse IFN-γ (430804, BioLegend). The assays were performed with 50 µL of supernatant separated from the T cell activation assay, according to the supplier’s protocol.
Animal experiment
As stated in our recent study,28 6–8 weeks old male BALB/c mice, male C57BL/6J mice, and NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt (NCG) mice were purchased from the Model Animal Research Center of Nanjing University. 6–8 weeks old male C57BL/6-Tg (TcraTcrb) 1100Mjb/J (OT-I) mice were purchased from the Jackson Laboratory. Animals were bred at the specific pathogen free (SPF) facility of the Medical School of Nanjing University.
For the in vivo subcutaneous (s.c.) murine xenograft model, 1×107 H22-CAR or Hepa 1–6 cells were s.c. inoculated into BALB/c mice. 1×106 MC38-CAR cells were s.c. inoculated into C57BL/6J mice. 2×106 LLC-CAR cells were s.c. inoculated into C57BL/6J mice. Tumor-bearing mice received three doses of PBS or OVs (5×108 IFU per dose, every other day) by intratumoral (i.t.) injection when the tumor burden reached approximately 100 mm3 as measured by calipers.
For the HCC ascites model, 2×106 H22-CAR cells were injected into the peritoneal cavity of BALB/c mice. When ascites formed, confirmed by peritoneal paracentesis, mice were randomly grouped and received three doses of PBS or OVs (5×107 IFU per dose, every other day) by i.p. injection.
To establish an orthotopic hepatocellular carcinoma model, 6–8 weeks old male BALB/c mice received laparotomy to expose the liver, then 1 × 10⁶ H22-CAR-Luc cells were injected under the liver capsule of the left lobe. To establish an orthotopic colon cancer model, 6–8 weeks old male C57BL/6J mice received laparotomy, then approximately a 1.0 mm diameter fragment of MC38-CAR-Luc tumor tissue dissected from s.c. tumor mass was implanted onto the cecum. On day 6 or 7 after tumor inoculation, mice received i.p. 150 mg/kg D-luciferin and were subsequently anesthetized using inhaled isoflurane, and bioluminescence imaging was performed using the AniView100 system. When orthotopic tumors were established, mice received a second laparotomy and i.t. injection of either 1.5×10⁹ IFU OVs or equal volume PBS. Tumor growth was further monitored through bioluminescent imaging on days 7 and 14 post-treatment. Survival was monitored every day. Tumor growth was analyzed using AniView software (V.1.00).
For human xenografts in NCG mice reconstituted with human lymphocytes, NCG mice were inoculated s.c. with 1×107 LM3 cells. After tumor appearance, mice were randomized on the basis of tumor size and intravenously injected with 1×107 human PBMCs. PBS or OVs (5×108 IFU per dose) were i.t. injected every other day for three doses.
For immune cell depletion, mice were given 200 µg of normal rat IgG or antibodies, including anti-CD4 antibody (Bio X Cell), anti-CD8 antibody (Bio X Cell) and anti-asialo GM1 antibody (Wako) by the i.p. route on the day when starting AdV-mSS treatment, and subsequently 200 µg every 3 days, two times. Clodronate liposomes (CL, 40 337ES10, Yeasen) were i.p. injected (150 µL of 7 mg/mL), and then 200 µl every 5 days. Depletion was verified by flow cytometry at the end of the experiment. Tumors were measured every other day using electronic calipers (KINOEE) and volume was calculated as follows: Tumor volume=length × width2/2. Animals were euthanized when the tumor volume exceeded 2000 mm3.
Flow cytometry
The s.c. 14-day H22-CAR or MC38-CAR-bearing mice were i.t. injected with PBS or OVs (5×108 IFU/dose) three times. 48 hours after treatment, tumors were digested in 2% RPMI 1640 medium containing 0.5 mg/mL collagenase type IV (17104019, Thermo Fisher Scientific) at 37°C for 1 hour. Cells were then passed through 70 µm cell strainers to obtain single-cell suspensions, followed by staining with a 1:100 diluted Fc blocker (anti-CD16/32) in 100 µL FACS buffer at 4°C for 20 min. Subsequently, cell suspensions were stained with the indicated antibodies (0.25 µg/106 cells) at 4°C for 30 min. Information on antibodies used in flow cytometry is provided in online supplemental table S2. For intracellular cytokine staining, the Fixation/Permeabilization Kit (BD Biosciences) was used. Flow cytometry was performed using BD Aria, BD Aria II Soap (BD Biosciences), Attune NxT (Thermo Fisher Scientific) and Beckman CytoFLEX LX (Beckman Coulter). The data were analyzed using FlowJo V.10 (Treestar).
RNA sequencing
For TAMs sequencing (GSE275905), the s.c. 14-day H22-OVA-bearing BALB/c mice were i.t. injected with PBS, 5×108 IFU AdV-Ctrl, or 5×108 IFU AdV-mSS three times. 48 hours after treatment, TAMs were isolated using anti-F4/80 MicroBeads (Miltenyi Biotec, 130-110-443), and total RNA from TAMs was extracted using RNeasy mini plus kit (74134, Qiagen) and stored at −80°C. Transcriptome RNA-seq analysis was performed using the Illumina NovaSeq 6000 sequencing platform (Majorbio).
In vitro T cell activation assay
The s.c. 14-day H22-OVA-bearing BALB/c mice were i.t. injected with PBS, 5×108 IFU AdV-Ctrl, or 5×108 IFU AdV-mSS three times. 48 hours after treatment, TAMs were isolated by anti-F4/80 MicroBeads (130-110-443, Miltenyi Biotec). Activated OT-I CD8+T cells were stained with 5 µM carboxyfluorescein succinimidyl ester (CFSE; 65-0850-84, eBioscience) working solution for 20 min at RT in the dark. TAMs were then co-cultured with CFSE-labeled OT-I T cells at a ratio of 2:1 in complete RPMI 1640 medium for 72 hours. Cells were collected, centrifuged, and the supernatant was separated. Subsequently, cells were stained with PerCP-Cy5.5 anti-CD8a antibody. T-cell proliferation was determined by flow cytometry using BD FACS Calibur (BD Biosciences).
Statistical analysis
Data analysis and visualization were conducted using Excel V.16.0 (Microsoft) and Prism 9 (GraphPad). Data were presented as mean±SD for in vitro experiments, or mean±SEM for in vivo experiments or individual values. For in vitro investigations, statistical analyses were assessed using either unpaired t-tests or one-way analysis of variance (ANOVA) with multiple comparison correction. For in vivo investigations, log-rank (Mantel-Cox) tests were used to evaluate survival curves, while two-way ANOVA with multiple comparison correction was used to compare tumor growth. p < 0.05 was considered statistically significant.
Results
Recombinant AdV-mSIRPα/Siglec-G enhances the phagocytosis of bone marrow-derived macrophages
To block CD47/SIRPα and Siglec-10/CD24, AdV was genetically engineered to encode a fusion protein containing both extracellular domains of murine SIRPα and Siglec-G (mouse ortholog of human siglec-10) (AdV-mSS). AdVs encoding either the single extracellular domain of murine SIRPα (AdV-mSIRPα) or murine Siglec-G (AdV-mSiglec-G) were developed as controls (figure 1A). Since the type 5 AdV infects mammalian cells via coxsackievirus and adenovirus receptors (CAR),29 and murine cell lines express low levels of CAR, we generated several CAR-expressing murine tumor cell lines, including H22-CAR, MC38-CAR, and LLC-CAR, to enhance AdV infectivity. Soluble mSIRPα, mSiglec-G, and the fusion protein mSIRPα/Siglec-G were detected in both supernatants and cell lysates from cells infected with the appropriate viruses (figure 1B). Moreover, the fusion protein did not impact viral infection and replication, as AdV-mSS showed comparable levels of replication and oncolysis to AdV-mSIRPα, AdV-mSiglec-G and AdV-Ctrl (online supplemental figure S1A,B).
Supplemental material
Using a competition binding experiment, we found that the supernatants from AdV-mSIRPα- or AdV-mSiglec-G-infected cells, competitively inhibited the binding of CD47 or CD24 antibodies, respectively. However, supernatant from AdV-mSS-infected cells affected the binding of both CD47 and CD24 antibodies (figure 1C), indicating a dual blockade of mSIRPα/Siglec-G against CD47 and CD24. We next investigated the impacts of mSIRPα/Siglec-G on the phagocytosis of BMDMs. Compared with supernatants from cells infected with AdV-Ctrl, AdV-mSIRPα, or AdV-mSiglec-G, supernatant from AdV-mSS-infected cells significantly enhanced the phagocytosis of BMDMs co-cultured with either H22-YFP (figure 1D) or MC38-YFP (figure 1E), accompanied by increased cytotoxicity (online supplemental figure S1C) and considerable production of TNF-α (online supplemental figure S1D). Additionally, BMDMs significantly enhanced the cytotoxicity of naïve T cells to tumor cells in the presence of supernatants obtained in AdV-mSS-infected cells (online supplemental figure S1C). Consistently, in an HCC ascites tumor model, AdV-mSS significantly facilitated TAM phagocytosis against H22-YFP-CAR cells compared with either AdV-Ctrl, AdV-mSIRPα, or AdV-mSiglec-G (figure 1F).
These results indicate that the fusion protein expressed by AdV-mSS exerts a dual blockade on CD47/SIRPα and CD24/Siglec-G pathways, thereby enhancing macrophage phagocytosis.
Targeted release of mSIRPα/Siglec-G by AdV-mSS significantly enhances antitumor efficacy in multiple solid tumor models
We investigated the antitumor efficacy of AdV-mSS in four xenograft models, including H22-CAR and Hepa 1–6 hepatocellular carcinomas, MC38-CAR colorectal carcinoma, and LLC-CAR Lewis lung carcinoma.
In the two HCC models (figure 2A), we observed substantial tumor growth inhibition and significantly increased survival in mice treated with AdV-mSIRPα, AdV-mSiglec-G, or AdV-mSS compared with AdV-Ctrl or PBS. AdV-mSS showed greater tumor inhibition, achieving a cure rate of 5/9 in the H22-CAR and 6/8 in the Hepa 1–6 models (figure 2B–E and online supplemental figure S2A–S3B). Similarly, AdV-mSS significantly enhanced tumor suppression and extended survival compared with AdV-mSIRPα or AdV-mSiglec-G in the MC38-CAR colon and LLC-CAR lung cancer models (figure 2F). However, no differences in tumor inhibition were found among AdV-Ctrl, AdV-mSIRPα, and AdV-mSiglec-G in the MC38-CAR colon cancer model (figure 2G,H and online supplemental figure S2C), and AdV-mSIRPα did not show improvement in the LLC lung cancer model (figure 2I,J and online supplemental figure S2D).

Intratumoral injection of AdV-mSS boosts antitumor efficacy in multiple solid tumor models. (A) Experimental setup to assess antitumor efficacy in subcutaneous hepatoma models. (B, C) Tumor progression (B) and Kaplan-Meier survival curves (C) of H22-CAR-bearing mice treated with PBS or OVs. (D, E) Tumor progression (D) and Kaplan-Meier survival curves (E) of Hepa 1–6 -bearing mice treated with PBS or OVs. (F) Experimental setup to assess antitumor efficacy in subcutaneous MC38-CAR or LLC-CAR models. (G–H) Tumor progression (G) and Kaplan-Meier survival curves (H) of MC38-CAR-bearing mice treated with PBS or OVs. (I–J) Tumor progression (I) and Kaplan-Meier survival curves (J) of LLC-CAR-bearing mice treated with PBS or OVs. (K–L) Experimental setup (K) and Kaplan-Meier survival curves (L) of mice bearing H22-CAR ascites hepatoma treated with PBS or OVs. The number of cured mice is indicated in brackets. The data are shown as the mean±SEM from one representative experiment out of at least two except for D and I, for which all mice are shown. P values were calculated using two-way ANOVA followed by Tukey’s multiple comparisons test (B, D, G and I) and the statistical evaluation of mouse survival was conducted using log-rank (Mantel-Cox) tests (C, E, H, J and L) (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-mSS, AdV-mSIRPα/Siglec-G; ANOVA, analysis of variance; CAR, coxsackievirus and adenovirus receptor; LLC, Lewis lung carcinoma; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OVs, oncolytic viruses; PBS, phosphate-buffered saline; s.c., subcutaneous; YFP, yellow fluorescent protein.
In addition to solid tumor models, we established an H22-CAR ascitic HCC model (figure 2K). Surprisingly, AdV-mSS treatment achieved a 100% cure rate, compared with cure rates of 3/7 in AdV-mSIRPα and 2/7 in AdV-mSiglec-G (figure 2L).
Taken together, the local release of the bispecific fusion protein mSIRPα/mSiglec-G by i.t. injection of AdV-mSS demonstrates more profound antitumor efficacy across a broader range of tumor subtypes.
Intratumoral administration of AdV-mSS reprograms TAMs by promoting antitumor polarization
We further analyzed the profiles of TAMs in tumor-bearing mice following i.t. administration of AdV-mSS. In the s.c. H22 model (figure 3A and online supplemental figure S3A), the infiltration of both CD11b+myeloid cells and TAMs was not increased by AdV-mSS (figure 3B and C). However, CD86+TAMs, which are antitumor subtypes, were significantly increased by AdV-mSS (figure 3D), while CD206+TAMs, which are protumor subtypes, were decreased (figure 3E). CD86 is a critical co-stimulatory molecule that interacts with CD28 on T cells, providing essential signals for T cell activation and proliferation during antigen presentation.30 The upregulation of CD86 on TAMs by AdV-mSS treatment suggests an enhanced ability to present antigens and facilitate effective T cell responses, thereby contributing to a more robust antitumor immune environment. Interestingly, these changes were not observed in peripheral (spleen) myeloid cells and macrophages (online supplemental figure S3B–3E). In line, the percentages of activated TAMs, including IFN-γ+ TAMs (figure 3F), TNF-α+ TAMs (figure 3G), and inducible nitric oxide synthase (iNOS)+TAMs (figure 3H), were significantly increased by AdV-mSS treatment compared with AdV-Ctrl treatment. Similar results were observed in the colorectal MC38-CAR tumor model (figure 3I–P).

Intratumoral administration of AdV-mSS promotes macrophage polarization towards antitumor phenotype. (A) Experimental setup of flow cytometry analysis for infiltrating immune cells in tumor and spleen samples from subcutaneous H22-CAR-bearing BALB/c mice after treatment. (B, C) Infiltration of myeloid cells (B; n=6) and macrophages (C; n=6) in H22-CAR hepatomas in mice treated with PBS or OVs. (D–H) Percentages of antitumoral macrophages—CD11b+F4/80+CD86+ cells (D; n=6), CD11b+F4/80+IFN-γ+ cells (F; n = 6), CD11b+F4/80+TNF-α+ cells (G; n =6), and CD11b+F4/80+iNOS+ cells (H; n =6)—and protumoral macrophages—CD11b+F4/80+CD206+ cells (E; n = 6)—in H22-CAR hepatomas of mice treated with PBS or OVs. (I) Experimental setup of flow cytometry analysis for infiltrating immune cells in tumor samples from subcutaneous MC38-CAR-bearing BALB/c mice after treatment. (J–L) Infiltration of myeloid cells (J; n=7) and macrophages (K; n=7) in MC38-CAR colorectal carcinoma of mice treated with PBS or OVs. (L–P) Percentages of antitumoral macrophages—CD11b+F4/80+CD86+ cells (L; n=7), CD11b+F4/80+IFN-γ+ cells (N; n=7), CD11b+F4/80+TNF-α+ cells (O; n=7), and CD11b+F4/80+iNOS+ cells (P; n=7)—and protumoral macrophages—CD11b+F4/80+CD206+ cells (M; n=7)—in MC38-CAR colorectal carcinoma of mice treated with PBS or OVs. Data are mean ± SD. P values were determined using one-way ANOVA with Tukey’s test (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-mSS, AdV-mSIRPα/Siglec-G; ANOVA, analysis of variance; CAR, coxsackievirus and adenovirus receptor; FACS, fluorescence-activated cell sorting; IFN, interferon; ifu, infectious unit; iNOS, inducible nitric oxide synthase; i.t., intratumoral; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OVs, oncolytic viruses; PBS, phosphate-buffered saline; s.c., subcutaneous; TNF, tumor necrosis factor.
We further evaluated the differential expression of TAMs isolated from H22 tumors treated with AdV-mSS or AdV-Ctrl. Transcriptome analysis revealed that TAMs exhibited significant overexpression of genes associated with phagosome, focal adhesion, and axon guidance pathways in the AdV-mSS group compared with the AdV-Ctrl group (online supplemental figure S3F,G). In addition, significant upregulation of several antitumor markers, including Tnfa, Il1b, and Irf7, and a decrease in protumor markers such as Il10, Arg1, Tgfb1, and Vegfa were observed in TAMs in the AdV-mSS group compared with AdV-Ctrl (online supplemental figure S3H).
Taken together, these results indicate that i.t. injection of AdV-mSS significantly switches TAMs to antitumor phenotypes within the TME.
Intratumoral administration of AdV-mSS strengthens CD8+ T cell-mediated antitumor immunity
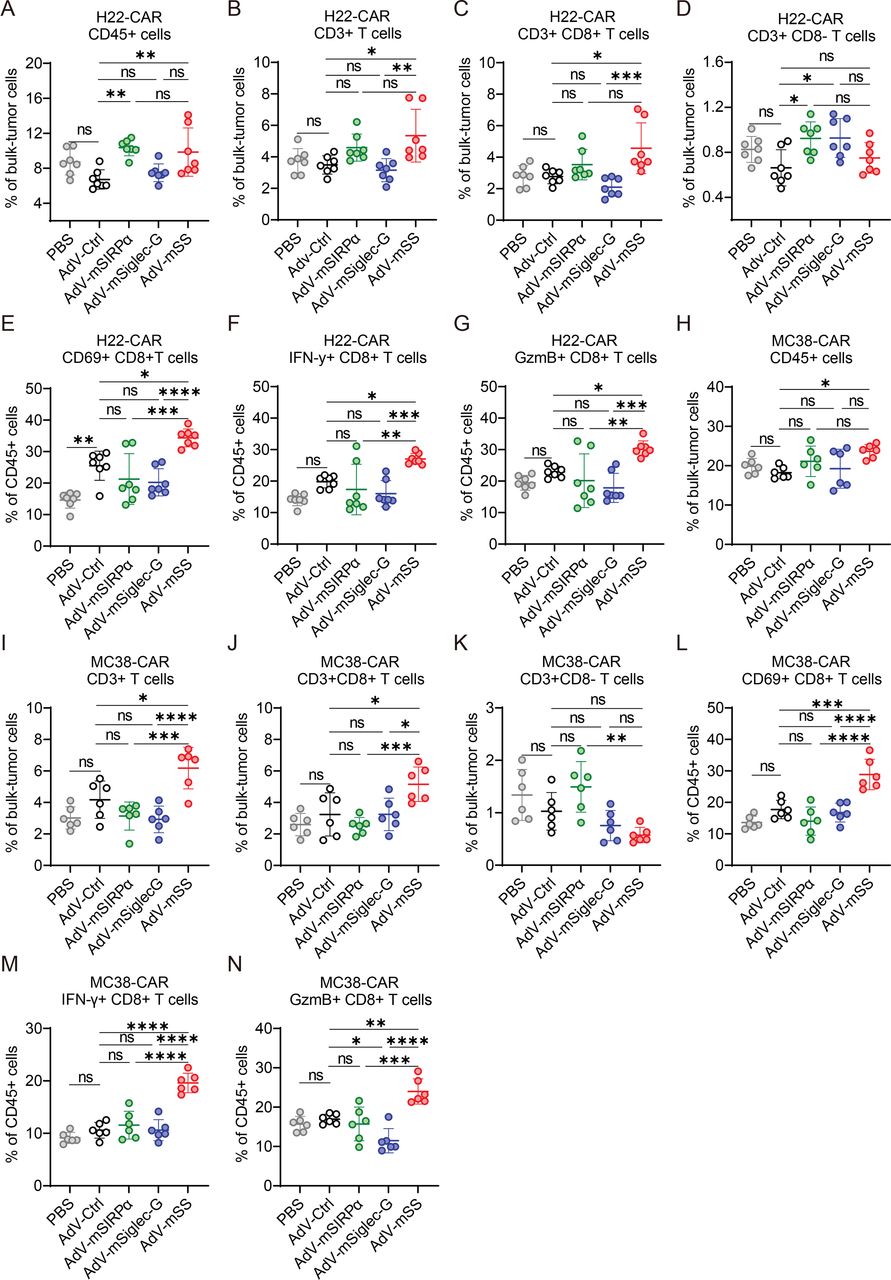
We next sought to determine whether the antitumor phenotype switch of TAMs alters the profiles of tumor-infiltrating T cells (TILs). In the s.c. H22 tumor model, infiltration of CD45+immune cells was significantly increased by AdV-mSS, AdV-mSIRPα, and AdV-mSiglec-G compared with AdV-Ctrl (figure 4A). The infiltration of CD3+T cells, specifically CD8+TILs, was significantly increased by AdV-mSS, whereas CD4+TILs were not significantly increased (figure 4B–D). Moreover, AdV-mSS treatment significantly activated CD8+TILs, as shown by an increased proportion of CD69+or IFN-γ+ CD8+ TILs, and a modest increase in GzmB+CD8+ TILs compared with AdV-Ctrl (figure 4E–G). The activated CD8+T cells were also observed in the spleens of tumor-bearing mice treated with AdV-mSS (online supplemental figure S4A–G).

Intratumoral delivery of AdV-mSS amplifies CD8+T cell-mediated antitumor response. (A–D) Infiltration of CD45+cells (A; n=7), T cells (B; n=7), CD8+T cells (C; n=7), and CD4+T cells (D; n=7) in H22-CAR hepatomas in mice treated with PBS or OVs. (E–G) Proportion of activated CD8+T cells, CD69+CD8+ T cells (E; n=7), IFN-γ+ CD8+ T cells (F; n=7), and GzmB+CD8+ T cells (G; n=7) in mice bearing H22-CAR treated with PBS or OVs. (H–K) Infiltration of CD45+cells (H; n=6), T cells (I; n=6), CD8+T cells (J; n=6), and CD4+T cells (K; n=6) in mice bearing MC38-CAR colon cancer treated with PBS or OVs. (L–N) Proportion of activated CD8+T cells, CD69+CD8+ T cells (L; n=6), IFN-γ+ CD8+ T cells (M; n=6), and GzmB+CD8+ T cells (N; n=6) in mice bearing MC38-CAR treated with PBS or OVs. The data are shown as the mean ± SD. P values were determined using one-way ANOVA with Tukey’s test (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-mSS, AdV-mSIRPα/Siglec-G; ANOVA, analysis of variance; CAR, coxsackievirus and adenovirus receptor; GzmB, Granzyme B; IFN, interferon; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OVs, oncolytic viruses; PBS, phosphate-buffered saline.
Similarly, in the s.c. colorectal MC38 tumor model, AdV-mSS treatment significantly increased the infiltration of CD3+and CD8+ TILs, but not CD4+TILs, compared with AdV-Ctrl (figure 4H–K). Consistently, AdV-mSS significantly improved the activation of CD8+TILs, as indicated by the extended proportions of CD69+CD8+ TILs, IFN-γ+ CD8+ TILs, and GzmB+CD8+ TILs (figure 4L–N).
These results indicate that AdV-mSS increases the infiltration of CD8+T cells and enhances their activity.
AdV-mSS enhances antitumor immune responses via the TAM-CD8+T cell axis
We further investigated the interaction between TAMs and CD8+T cells. Compared with AdV-Ctrl, AdV-mSS treatment significantly increased the expression of MHC-II and MHC-I in TAMs, which are indispensable for T cell activation (figure 5A). To further evaluate if TAMs enhance the cytotoxicity of tumor-specific CTLs, mice bearing H22 cells stably expressing ovalbumin (H22-OVA) received i.t. injections of AdV-mSS, then the TAMs isolated and co-cultured with CFSE-labeled OT-I T cells, which can be specifically activated by the presentation of OVA-related antigens. Indeed, TAMs obtained from H22-OVA tumors treated with AdV-mSS markedly induced the proliferation of OT-I cells (figure 5B) and IFN-γ production in OT-I cells (figure 5C). Similarly, increased MHC-II and MHC-I expression were observed in TAMs from MC38-CAR tumors treated with AdV-mSS compared with AdV-Ctrl treatment (online supplemental figure S4H). These results indicate that AdV-mSS enhances the antigen presentation capability of TAMs, thereby improving T cell proliferation and activation.

AdV-mSS modulates the TAM-CD8+T cell axis to strengthen antitumor responses (A) Expression of antigen-presenting MHC-II and MHC-I molecules on TAMs in H22-CAR tumors from mice treated with PBS or OVs. n=8. (B) Experimental setup, representative plots and percentages of CFSE-labeled OT-I CD8+T cells co-cultured with TAMs isolated from H22-OVA tumor-bearing mice treated with PBS, AdV-Ctrl, or AdV-mSS. n=3. (C) Cytokine IFN-γ production from OT-I CD8+T cells co-cultured with TAMs isolated from H22-OVA tumor-bearing mice treated with PBS, AdV-Ctrl, or AdV-mSS, three mice each group. (D) Experimental setup and tumor progression of subcutaneous H22-CAR-bearing BALB/c mice treated with AdV-mSS following immune cell depletion, five mice each group. (E) Experimental setup of C57BL/6J mice bearing orthotopic MC38-CAR-Luc tumors following intratumoral injection of PBS or Ovs as indicated, (F) tumor growth monitoring, and Kaplan-Meier survival curves, five mice each group. (G) Experimental setup of BALB/c mice bearing orthotopic H22-CAR-Luc tumors following intratumoral injection of either PBS, AdV-Ctrl, or AdV-mSS, (H) tumor growth monitoring, and Kaplan-Meier survival curves, five mice each group. The data are shown as mean±SD for A–C or the mean±SEM for D, F and H. P values were analyzed by one-way ANOVA with Tukey’s test for A–C, and two-way ANOVA with Tukey’s test for D, F and H (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-mSS, AdV-mSIRPα/Siglec-G; ANOVA, analysis of variance; CAR, coxsackievirus and adenovirus receptor; CFSE, carboxyfluorescein succinimidyl ester; FITC, fluorescein Isothiocyanate;IFN, interferon; ifu, infectious unit; i.p., intraperitoneal; i.t., intratumoral; Luc, luciferase; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OVA, ovalbumin; OVs, oncolytic viruses; PBS, phosphate-buffered saline; s.c., subcutaneous; TAMs, tumor-associated macrophages.
We then investigated which subpopulations of tumor-infiltrating immune cells contributed to AdV-mSS-mediated antitumor effects. In a s.c. H22-CAR HCC model, CD4+T cells, CD8+T cells, NK cells, or macrophages were depleted by respective monoclonal antibodies (figure 5D). Flow cytometry was performed to confirm depletion efficiency (online supplemental figure S4I). The antitumor effects of AdV-mSS were abrogated in mice that received either depletion of macrophages or CD8+T cells, but not in mice with CD4+T or NK cell depletion (figure 5D). These results suggest that both macrophages and CD8+T cells are involved in AdV-mSS-enhanced antitumor effects.
Given that TME, specifically how the tumor interacts with surrounding healthy tissue, is crucial in tumor development and impacts the outcomes of immunotherapies. Therefore, we further established an orthotopic MC38 colon cancer model (figure 5E), and found that i.t. injection of AdV-mSS significantly enhanced tumor suppression and extended survival compared with either AdV-Ctrl, AdV-mSIRPα or AdV-mSiglec-G, achieving a cure rate of 3/5 (figure 5F). Similarly, in another orthotopic H22 hepatocellular carcinoma model (figure 5G), i.t. injection of AdV-mSS significantly inhibited tumor progression and ascites formation, leading to prolonged survival, achieving a cure rate of 2/5 (figure 5H). These results further validate the conclusions we obtained in the s.c. tumor models.
We also evaluated the safety profile of AdV-mSS in treated mice. Similar levels of viral replication in tumors were found between AdV-mSS and the corresponding AdVs (online supplemental figure S5A), while no viruses were detected in other organs after AdV injection (online supplemental figure S5B). AdV-derived proteins were detected in tumor tissues but not in the blood (online supplemental figure S5C). No tissue damage was observed in organs including the brain, cerebellum, heart, lung, liver, spleen, kidney, small intestine, and muscle after AdV-mSS treatment (online supplemental figure S5D).
Taken together, our findings suggest that AdV-mSS effectively reprograms TAMs towards antitumor phenotypes, thereby enhancing the tumor-killing ability of CD8+T cells.
Oncolytic AdV-huSS inhibits the progression of LM3 hepatocellular carcinoma in humanized mice
Finally, we constructed a recombinant oncolytic AdV expressing human SIRPα/Siglec-10 (AdV-huSS) (figure 6A). The fusion protein expressed by AdV-huSS specifically bound to CD47 and CD24 on the surface of the human LM3 hepatocellular carcinoma cell line (figure 6B). Human monocyte-derived macrophages exhibited enhanced phagocytosis in the presence of the supernatant from AdV-huSS-infected cells (figure 6C), accompanied by increased macrophage cytotoxicity and TNF-α secretion (figure 6D). In humanized NCG mice bearing s.c. LM3 hepatocellular carcinoma (figure 6E), AdV-huSS significantly inhibited tumor growth compared with PBS or AdV-Ctrl treatment (figure 6F), and no significant weight loss was observed (figure 6G). These results demonstrate that AdV-huSS possesses potent antitumor activity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Oncolytic AdV-huSS suppresses LM3 hepatocellular carcinoma growth in a humanized mouse model. (A) Expression of human Siglec-10 in OV-infected 293T cells was detected by western blot analysis to determine the concentrations of soluble huSIRPα/Siglec-G proteins. Culture supernatants and cell lysates were obtained. n=3 independent experiments. (B) Competitive flow cytometry was used to evaluate the binding capacity of fusion proteins to human hepatocellular carcinoma LM3 cells. Conjugated anti-human CD24 or anti-human CD47 antibodies competed with fusion proteins extracted from the supernatants of OV-infected 293T cells for binding to CD24 or CD47 on the surface of tumor cells. The results were assessed by flow cytometry. n=3 independent experiments. (C) Quantification of in vitro phagocytosis against LM3-YFP cells by human MDMs with supernatants from OV-infected 293T cells. The percentage of phagocytosis against LM3-YFP cells (CD11b+YFP+ cells) was calculated by flow cytometry. n=3 independent experiments. (D) In vitro cytotoxicity and TNF-α production of huMDMs co-cultured with LM3-Luc cells in supernatant from OV-infected 293T cells. n=3 independent experiments. (E–G) Experimental setup (E), tumor progression (F) and body weights (G) of human LM3 subcutaneous model in NSG mice treated with PBS, AdV-Ctrl or AdV-huSS after adoptive transfer of allogeneic human PBMCs. n=5 mice. The data are the mean±SD. P values were determined using two-way ANOVA with Sidak’s s test (B), one-way ANOVA with Tukey’s test (C–D) and two-way ANOVA with Tukey’s test (F) (ns, not significant; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). AdV, adenovirus; AdV-Ctrl, AdV-control; AdV-huSS, AdV expressing human SIRPα/Siglec-10; ANOVA, analysis of variance; FITC, fluorescein Isothiocyanate; huMDMs, human monocyte-derived macrophages; huPBMCs,human peripheral blood mononuclear cells; i.t., intratumoral; i.v., intravenous; Luc, luciferase; mSiglec-10, murine sialic acid-binding immunoglobulin-like lectin 10; mSIRPα, murine signal regulatory protein alpha; OV, oncolytic virus; PBS, phosphate-buffered saline; s.c., subcutaneous; TNF, tumor necrosis factor; YFP, yellow fluorescent protein.
Discussion
In this study, we developed a recombinant AdV-SS encoding the bispecific fusion protein SIRPα/Siglec-10, serving as a local delivery vector to block both immune checkpoints CD47 and CD24 in the TME. The i.t. administration of AdV-SS reprogrammed TAMs towards antitumor phenotypes, resulting in enhanced phagocytosis and MHC-I/II expression, which significantly improved CD8+T cell-mediated antitumor effects across a series of tumors.
Systemic administration of CD47-targeted therapies, such as monoclonal antibodies and recombinant SIRPα-Fc fusion proteins, has displayed potential antitumor activity. CD47/SIRPα-based bispecific antibodies and fusion proteins have been developed to enhance tumor-specific targeting and reduce off-target effects.31 Recently, a study developed a bispecific fusion protein targeting both CD47 and CD24, which demonstrated superior tumor inhibition compared with either CD47 or CD24 monotherapy.32 Nevertheless, systemic administration of CD47/SIRPα-based bispecific antibodies hardly circumvent the clinical obstacles associated with the elimination of healthy CD47-expressing cells, including red blood cells and platelets.33–36 The i.t. injection of AdV-SS sophistically overcomes the off-target hurdle by local generation and release of the bispecific SIRPα/Siglec-10 in the TME.
Dual blockade of CD47/CD24 by AdV-SS significantly invigorated the activity and phagocytosis of TAMs, leading to better therapeutic outcomes than inhibition of either CD47 or CD24 alone across a series of tumor models. Beyond the improved antitumor activities of TAMs, AdV-mSS also significantly increased tumor-infiltrating CD8+T cells and their immune activity. These effects were likely associated with the upregulated activity of TAMs, including increased expression of chemokines. Additionally, upregulated MHC-I and MHC-II on TAMs by AdV-mSS promoted crosstalk between TAMs and CD8+T cells, leading to enhanced activation of tumor-infiltrating CD8+T cells. Indeed, our results showed that the TAM-CD8+T cell axis was involved in AdV-mSS-induced antitumor effects. Therefore, the distinct subtypes of immune microenvironments are reasonably expected to exhibit diverse responses to our present therapeutic strategy. Predictably, malignancies in which the TME riched in TAMs and T cells might demonstrate heightened sensitivity to AdV-mSS. In addition, AdV-mSS might be suitable for combination with other immunotherapies, such as adoptive T cell therapy (eg, CAR-T or TILs) and immune checkpoint blockades (ICBs), to further improve the therapeutic benefits.
To our surprise, we observed a 100% cure rate in ascitic HCC mouse model, suggesting that AdV-SS may be particularly suitable for the treatment of malignant ascites, a significant challenge in clinical therapy. In this study, we also investigated the antitumor efficacy of a recombinant AdV encoding the human SIRPα/Siglec-10 fusion protein and observed significantly improved antitumor outcomes in a humanized HCC mouse model. This finding warrants further preclinical investigation and clinical evaluations in patients with advanced malignancies, including malignant ascites.
Our study revealed that AdV-SS not only reinvigorates the antitumor activity of TAMs but also induces TAM-CD8+T cell axis-mediated antitumor effects. Moreover, i.t. administration of AdV-SS to achieve local delivery of the dual blocker SIRPα/Siglec-10 may bypass the off-target risks associated with systemic delivery of CD47/SIRPα-based bispecific antibodies. Our work provides a potentially safe and effective antitumor candidate for clinical cancer therapy.
Data availability statement
Data are available upon reasonable request. The RNA-seq data used in this study have been deposited at the Gene Expression Omnibus under accession codes (SuperSeries GEO accession, GSE275905). The remaining data generated or analyzed during this study are available within the article and Supplementary Information. Source data are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The handling of mice and experimental procedures were conducted in accordance with national and institutional guidelines for animal care and were approved by the Science and Technology Ethics Committee of Nanjing University (D2403019).
References
Footnotes
Contributors JW conceived the study. JW and JD supervised the project. JW, JD, and YZ designed the experiments. YZ, BH, PZ, MWu, MWe, and CX performed the experiments. YZ analyzed the results. JW, JD, and YZ drafted the manuscript. All authors critically reviewed and approved the manuscript. The guarantor of the study is JW.
Funding This study was supported by the National Natural Science Foundation of China (82273261 to JW), Nanjing University (0214/151130 to JW), by the State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University (ZZYJ-202401 to JW), and by the Postgraduate Research and Practice Innovation Program of Jiangsu Province (KYCX24_0285 to YZ).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.